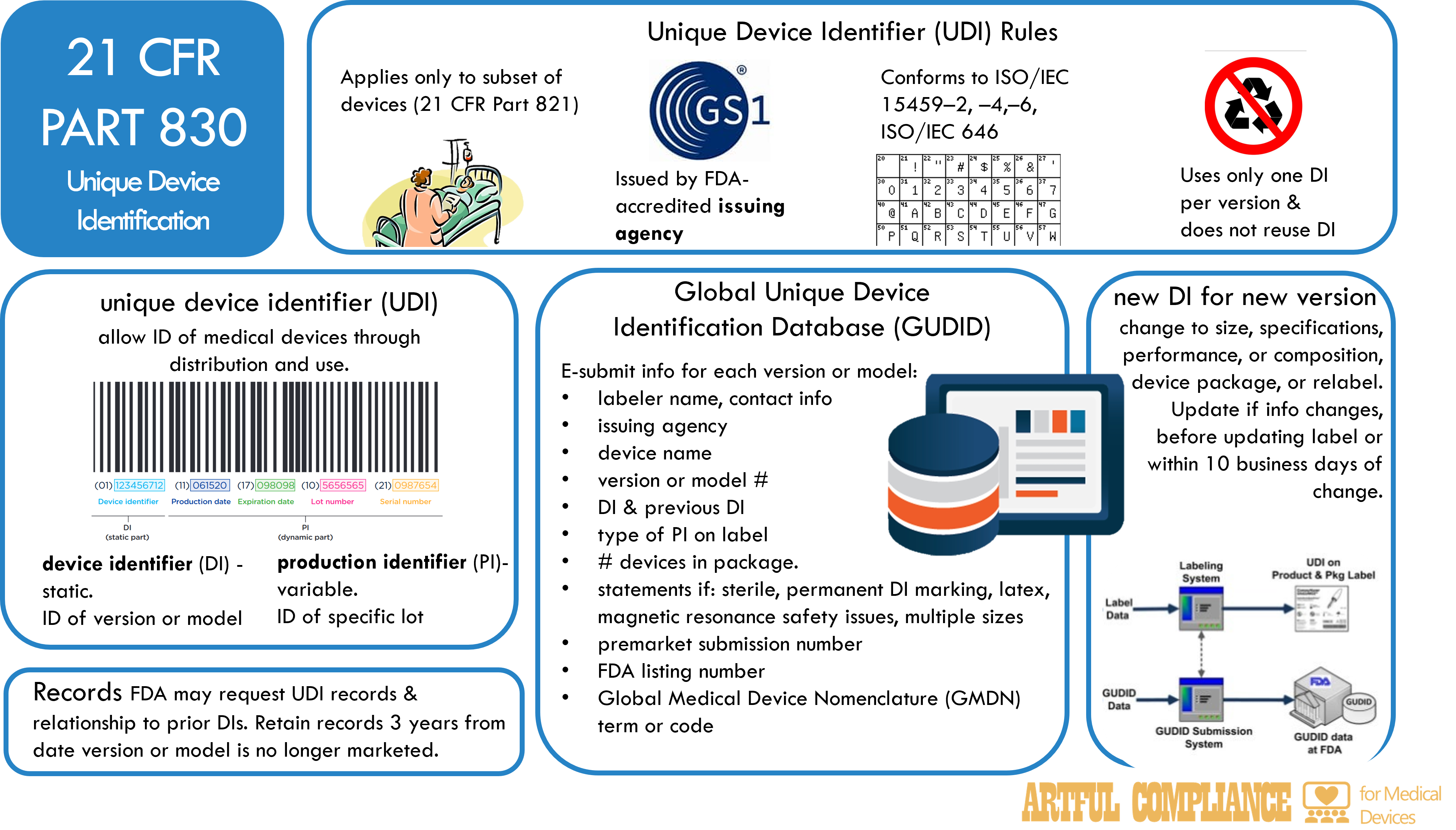
The FDA requires certain medical devices to have a unique device identifier (UDI) that can be used to identify and track them throughout their distribution and use. A UDI is a code that consists of two parts: a device identifier (DI) and a production identifier (PI). The DI identifies the specific version or model of a device, and the PI identifies the unit of device production, such as the lot or batch number, the serial number, the expiration date, or the date of manufacture. The UDI must be displayed on the device label and package, and in some cases, on the device itself. The purpose is to improve patient safety by making device recalls and postmarket surveillance easier.
UDI Applies to Only a Subset of Devices
The UDI system applies to a subset of devices that are classified by the FDA according to their risk level (see 21 CFR Part 821 for the list of devices and compliance dates).
Issuing UDIs
The UDI must be issued by an FDA-accredited issuing agency, such as GS1, and conform to international standards for data structure and character set. The UDI must use only one DI per version or model of a device, and must not reuse a DI that has been previously assigned to another device.
Global Unique Device Identification Database (GUDID)
The FDA also maintains a Global Unique Device Identification Database (GUDID) that stores the UDI and other information about each device. The GUDID allows the public and health care professionals to access information about medical devices. The labeler of a device, which is usually the manufacturer, is responsible for electronically submitting the UDI and other information to the GUDID no later than the date the label is placed on the device, or the date the device is released for commercial distribution. The labeler must also update the GUDID if any information changes, or if a new DI is required for a device.
The information that must be submitted to the GUDID for each version or model of a device includes:
- The labeler’s name and contact information, and the name of the issuing agency
- The DI, and any previous DI that was assigned to the device
- The device name, and the version or model number
- Any applicable statements that indicate if the device is sterile, has a permanent marking of the DI, contains natural rubber latex, has safety issues if exposed to magnetic resonance, or has multiple sizes
- The type of PI that is displayed on the device label
- The premarket submission number, if any, that was assigned by the FDA
- The FDA listing number that identifies the device
- The Global Medical Device Nomenclature (GMDN) term or code that describes the device
- The number of devices contained in each package
The FDA may grant a waiver or an exception from the UDI requirements for certain devices, or allow voluntary submission of UDI information for devices that are not subject to the requirements.
Significant Changes to Devices Require a New UDI
A new DI is required for a device if there is a change in the device’s specifications, performance, size, or composition that could affect its safety or effectiveness, or if there is a change in the device’s package or labeling. The labeler must notify the FDA of any changes that require a new DI, and update the GUDID accordingly.
Corrections to UDI Data
The FDA may review the GUDID data and request corrections or explanations from the labeler if the data is incorrect or misleading. The FDA may also delete or modify the data if necessary.
Records
The labeler must keep records of all UDIs and the relationship of prior and current DIs for each device, and retain the records for three years after the device is no longer marketed. The FDA may inspect the records and request copies as part of its regulatory oversight.
Recent Comments