00085 VALID Act Pre-Market Review Application
From Valid Act SEC. 587B. Premarket review.
“(c) Application.—
“(1) FILING.—Any person may file with the Secretary an application for premarket approval of an in vitro clinical test.
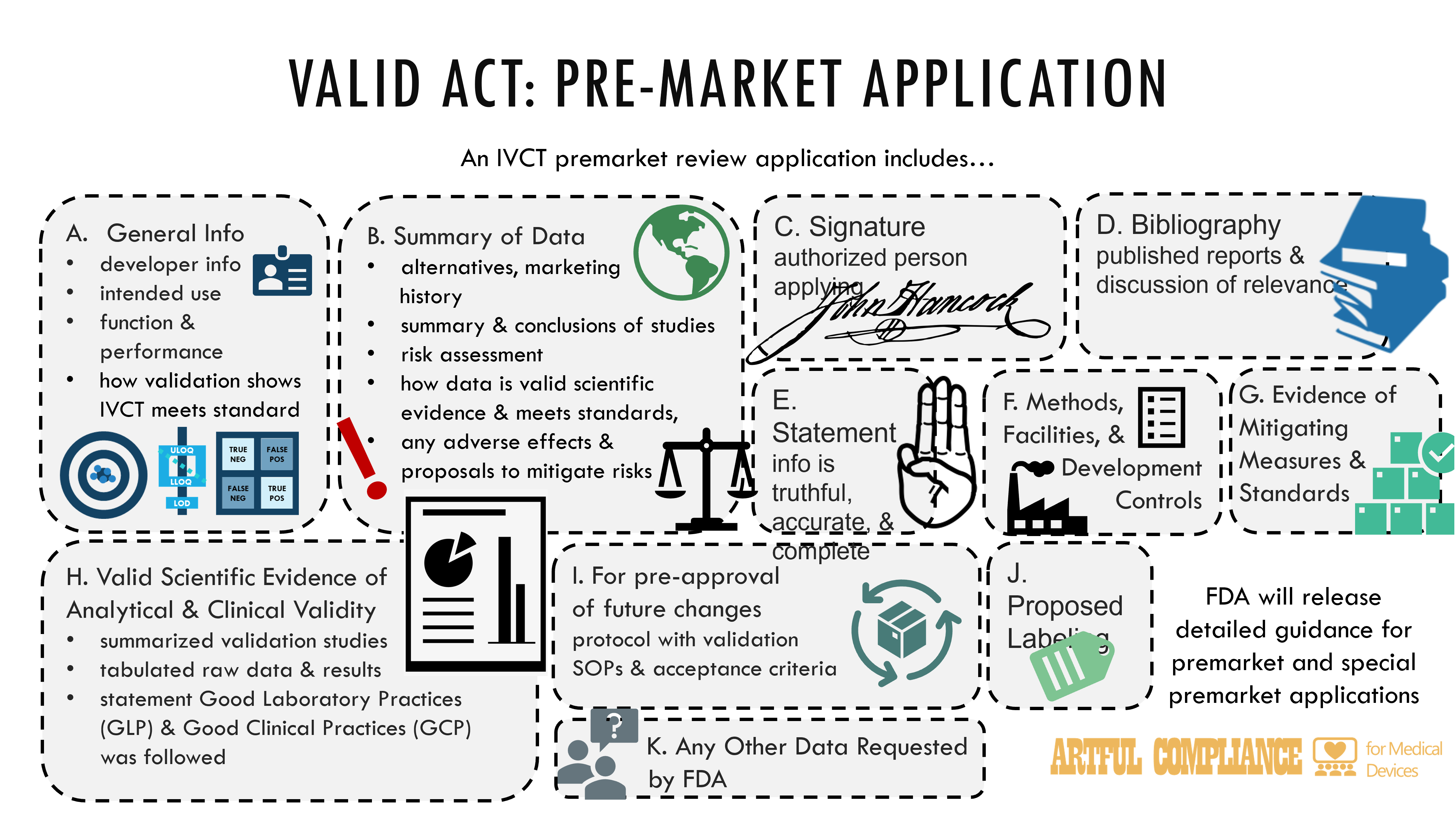
“(2) APPLICATION CONTENT.—An application submitted under paragraph (1) with respect to an in vitro clinical test shall include the following, in such format as the Secretary specifies:
“(A) General information regarding the in vitro clinical test, including—
“(i) the name and address of the applicant;
“(ii) the table of contents for the application and the identification of the information the applicant claims as trade secret or confidential commercial or financial information;
“(iii) a description of the test’s intended use;
“(iv) an explanation regarding test function and any significant performance characteristics; and
“(v) an explanation of how the development and validation activities support the test meeting the applicable standard.
“(B) A summary of the data and information in the application for the in vitro clinical test, including—
“(i) a brief description of any existing alternative practices or procedures for diagnosing the disease or condition for which the in vitro clinical test is intended, as applicable;
“(ii) a brief description of the foreign and domestic marketing history of the test, if any, including a list of all countries in which the test has been marketed and a list of all countries in which the test has been withdrawn from marketing for any reason related to the applicable standard of the in vitro clinical test, if known by the applicant;
“(iii) a summary of the any studies submitted for such test, including a description of the objective of the study, a description of the experimental design of the study, a brief description of how the data were collected and analyzed, a brief description of the results of the technical data submitted, and a brief description of any nonclinical or clinical studies;
“(iv) a risk assessment of the test; and
“(v) conclusions drawn from any studies described in clause (iii), including a discussion demonstrating that the data and information in the application constitute valid scientific evidence and meet the applicable standard under section 587(2), an explanation of how the development and validation activities, as applicable, support that the test meets the applicable standard under section 587(2), and a discussion of any adverse effects of the test on health and proposals to mitigate those risks, if any.
“(C) The signature of the person filing the premarket application or an authorized representative.
“(D) A bibliography of all published reports reasonably known to the applicant related to such test and a discussion of data and information relevant to the evaluation of the applicable standard that may be met by such test.
“(E) A statement that the applicant believes to the best of the applicant’s knowledge that all data and information submitted to the Secretary are truthful and accurate and that no material fact has been omitted in the application.
“(F) Except as provided under subsection (d), applicable information regarding the methods used in, or the facilities or controls used for, the development of the test to demonstrate compliance with the applicable quality requirements under section 587J.
“(G) Information demonstrating compliance with any relevant—
“(i) mitigating measures under section 587E; and
“(ii) standards established or recognized under section 514 prior to the date of enactment of the Verifying Accurate Leading-edge IVCT Development Act of 2021, or, after applicable standards are established or recognized under section 587Q, with such standards.
“(H) Valid scientific evidence to support analytical and clinical validity of the test, which shall include—
“(i) summary information for all supporting validation studies performed;
“(ii) raw data, such as tabulations of data and results as required under section 814.20(b)(6)(ii) of title 21, Code of Federal Regulations (or any successor regulations);
“(iii) for nonclinical laboratory studies involving the test, a statement that studies were conducted in compliance with applicable good laboratory practices; and
“(iv) for investigations involving human subjects, statements that any clinical investigation involving human subjects was conducted in compliance with applicable—
“(I) institutional review board regulations;
“(II) informed consent regulations; and
“(III) investigational use requirements in section 587R.
“(I) To the extent the application seeks authorization to make modifications to the test within the scope of the approval, a change protocol that includes validation procedures and acceptance criteria for anticipated modifications that could be made to the test within the scope of the approval.
“(J) Proposed labeling, in accordance with the requirements of section 587K.
“(K) Such other data or information as the Secretary may require in accordance with the least burdensome requirements of subsection (j).
“(3) GUIDANCE FOR PREMARKET AND SPECIAL PREMARKET APPLICATIONS.—In accordance with section 5 of the Verifying Accurate Leading-edge IVCT Development Act of 2021, the Secretary shall issue draft guidance detailing the information to be provided in a premarket application and special premarket application under this section. The Secretary shall issue final guidance not later than 90 calendar days after the close of the comment period for such guidance.
Recent Comments