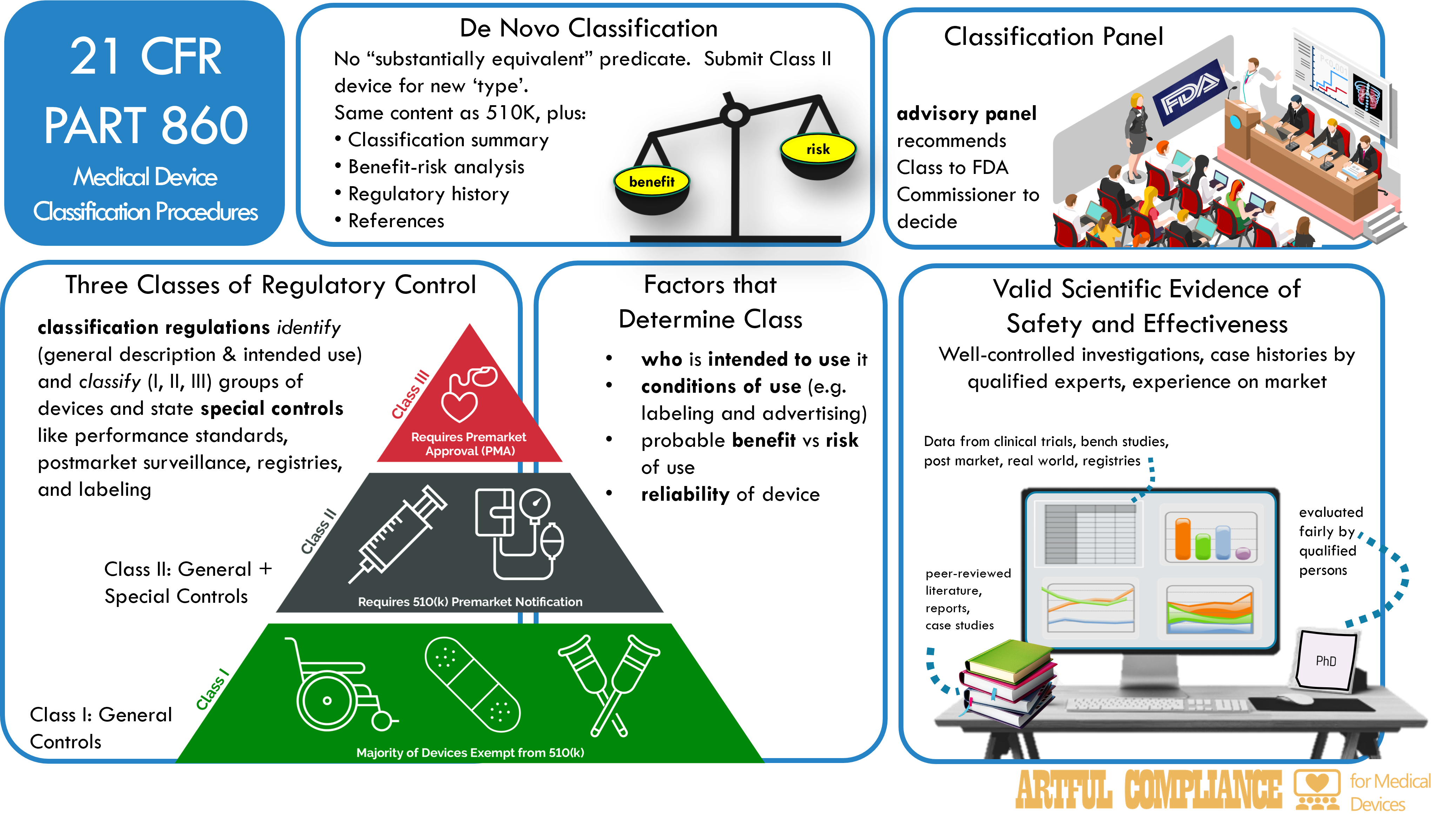
Three Classes of Regulatory Control
The FDA classifies devices into one of three classes based on their risk and regulatory oversight.
| Class | Risk Level | Controls | Examples |
|---|---|---|---|
| I | Low | General | Bandages, tongue depressors, dental floss |
| II | Moderate | Special + General, premarket notification and clearance | Pregnancy tests, blood glucose meters, infusion pumps |
| III | High | General, premarket approval | Implants, pacemakers, artificial heart valves, companion diagnostics |
- general controls include registration, listing, labeling, good manufacturing practices, and adverse event reporting.
- special controls include performance standards, postmarket surveillance, patient registries, special labeling, or guidance documents.
Classification Panel
A classification panel is an advisory committee of experts that makes recommendations to the FDA on the classification and reclassification of devices. The FDA has 16 classification panels, each covering a major medical specialty or device type such as Anesthesiology, Cardiovascular, and Pathology.
To simplify regulations for special controls, devices are grouped into generic types of devices, which have similar features and require similar regulatory controls to ensure their safety and effectiveness. Classification panels help the FDA develop a classification regulation for each type of device (see 21 CFR parts 862 through 892). A panel may also recommend whether a class I or II device should be exempt from some or all general or special controls.
Factors that Determine Classification
The FDA classifies a device based on the following factors:
- Intended users: The persons who will use the device
- Indications and conditions of use: How, when, and where the device will be used, including labeling and advertising
- Technological characteristics: The design, material, and performance of the device
- Benefit-risk profile: The potential benefits and harms of using the device
- Regulatory necessity: The level of regulation needed to ensure safety and effectiveness
- Regulatory alternatives: The existing options for regulating the device
- Reliability: The consistency and accuracy of the device
- Novelty: The degree of innovation and uniqueness of the device
- Availability of alternatives: The presence or absence of other devices that serve the same or similar purposes
- Public health need: The importance and urgency of addressing the medical problem that the device aims to solve
- Health care system impact: The effect of the device on the cost, quality, and accessibility of health care
Evidence for Classification
For devices that require premarket approval or clearance, the FDA requires valid scientific evidence of safety and effectiveness to classify the device. This includes:
- Well-controlled investigations: Studies that follow rigorous scientific principles and use appropriate control groups and methods
- Objectives and methods: The study plan and report should clearly state the goals, the selection and assignment of subjects, the observation and measurement methods, and the comparison and analysis of results.
- Control groups: The study should use a suitable control group that minimizes bias and allows quantitative evaluation. The study should explain the type and methods of the control group and the degree and methods of blinding, if any.
- Test device standardization: The study should use a test device that is consistent in its composition, design, and performance to ensure the reliability of the results.
- Partially controlled studies: Studies that use some but not all elements of well-controlled investigations
- Uncontrolled studies: Studies and trials that do not use control groups but have other means of ensuring validity and reliability
- Qualified case histories: Well-documented reports of clinical experience by experts
- Significant human experience: Reports of widespread and consistent use of a marketed device
- Nonclinical methods: Laboratory tests, animal studies, or analytical studies for in vitro diagnostic devices, when required, to assess the safety of the device
Note that isolated case reports, random experience, reports lacking details, and unsubstantiated opinions are NOT valid scientific evidence.
How the FDA Evaluates Evidence
The FDA evaluates the quality, quantity, relevance, and applicability of the evidence to the device and its intended use. The evidence must:
- Demonstrate safety: Show that the device does not pose unreasonable risk of illness or injury for its intended uses and conditions of use, based on valid scientific evidence
- Demonstrate effectiveness: A device is effective if it produces clinically significant results for most of the target population, under the intended uses and conditions of use, with proper instructions and warnings.
- Provide directions and warnings: Include adequate instructions and cautions to prevent unsafe use of the device
When submitting evidence, the sponsor must:
- Comply with any reports or information requests related to the classification of the device and its safety and effectiveness, including the reason, description, time, form, and manner of the report or information.
- Incorporate by reference any required information that has been previously submitted to the relevant Center.
De Novo Classification
The de novo classification process is for novel devices that are not similar to any existing device or are automatically classified as Class III. The FDA can classify such devices as Class I or II based on their risk and regulation level.
The process begins with a De Novo request from the device sponsor, which includes the device description, proposed classification, benefits and risks summary, scientific evidence summary, and bibliography. The FDA will make a decision within 150 days, and may ask for more information, consult experts or panels, or inspect the device.
The sponsor can withdraw the request before the FDA issues an order, but will not get a refund of the de novo fee. The FDA will grant or decline the request based on the evidence and risk, and will issue an order, publish it in the Federal Register, and update the database. De Novo submissions are confidential until granted.
Reclassification
Reclassification is changing the class of a device. A reclassification petition from an interested person or a device manufacturer or importer contains the type, action, basis, reasons, and data for reclassification. The classification panel will recommend the appropriate class before the FDA or the Commissioner issues an order to reclassify the device. There are five types of reclassification processes, depending on the situation and the law. They are:
- Based on new or existing information on its safety and effectiveness.
- Based on new performance standard or information, requiring premarket approval for a Class I or II device.
- Requiring premarket approval for a Class III device subject to premarket notification.
- Reclassifying a device without a predicate device (De Novo request).
- Reclassifying a product that was a drug or biological product before 1976. Each process starts with a petition or notification from the FDA, with panel consultation and public comment.
Recent Comments