Postmarket surveillance (PMS) is the process of monitoring the safety and effectiveness of medical devices after they have been approved or cleared for marketing by the FDA. PMS can help identify unforeseen adverse events, the actual rate of anticipated adverse events, or other information necessary to protect the public health when using a medical device.
Applicability
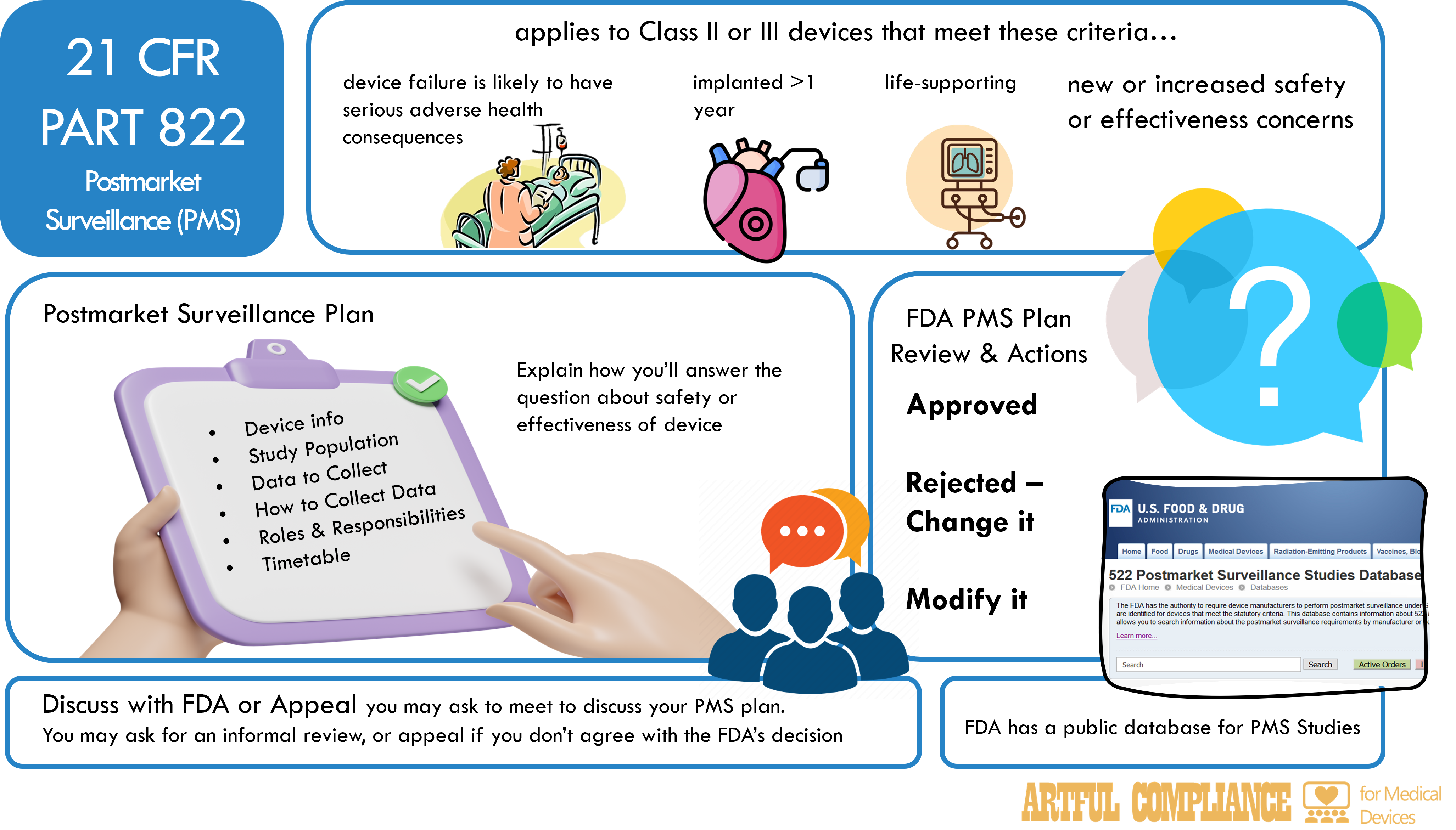
Not all medical devices require PMS. If PMS is needed, the FDA will let you know by issuing an order that specifies the reason for the surveillance, the type of surveillance, and the timeframe for submitting and completing the surveillance plan. The FDA may order PMS for any device that meets one or more of the following criteria.
- The failure of the device would be reasonably likely to have serious adverse health consequences.
- The device is intended to be implanted in the human body for more than one year.
- The device is intended to be a life-sustaining or life-supporting device used outside a device user facility.
- The device is new or modified and raises new or increased safety or effectiveness concerns.
PMS orders are mostly for Class III devices. The FDA has a database that shows all active and completed PMS orders here: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pss.cfm
Postmarket Surveillance Plan
The manufacturer must submit a postmarket surveillance plan to the FDA within 30 days of receiving the order, unless otherwise specified. The plan must be scientifically sound and include the following elements:
- A statement of the postmarket surveillance question or issue and an explanation of how the data or other information to be collected will address it.
- A description of the device, including the indications for use, the marketing history, and any relevant information from premarket testing or postmarket studies.
- A description of the population to be studied, including the number of patients, the selection criteria, the enrollment procedure, and the follow-up schedule.
- A description of the data or other information to be collected, including the sources, the methods, the variables, the endpoints, and the analysis plan.
- A description of the surveillance methods, including the study design, the data collection procedure, the quality control measures, and the data management system.
- A description of the organizational structure, including the roles and responsibilities of the manufacturer, the investigators, the contractors, and the institutional review boards (IRBs).
- A timetable for conducting the surveillance, including the milestones, the interim reports, and the final report.
IRB Approval
Because a PMS study is also a human study, the plan must be reviewed and approved by an IRB that meets the requirements of 21 CFR Part 56. The manufacturer must also obtain informed consent from the patients who participate in the surveillance, unless the FDA or the IRB waives this requirement.
FDA Review and Actions
The FDA will review the postmarket surveillance plan and (typically) within 60 days either approve, disapprove, or request modifications to the plan.
- If approved, the plan must be followed and you must submit periodic reports to the FDA. You must also notify the FDA of any changes to the plan and obtain the FDA’s approval before making the changes.
- If disapproved, you typically must submit a new plan within 15 days of receiving the notice of disapproval. The FDA may take regulatory actions, such as administrative detention, seizure, injunction, or civil money penalties, if you don’t submit a new plan or submit an inadequate plan.
- If the FDA requests modifications to the plan, you must typically revise and submit a changed plan within 15 days. Again, the FDA will give a decision within 60 days.
You can request a meeting with the FDA to discuss the plan or the FDA’s decision. There is also the option for an informal review or a formal appeal of the decision.
The FDA may terminate the postmarket surveillance order if they determine it is no longer needed to protect the public health.
Recent Comments