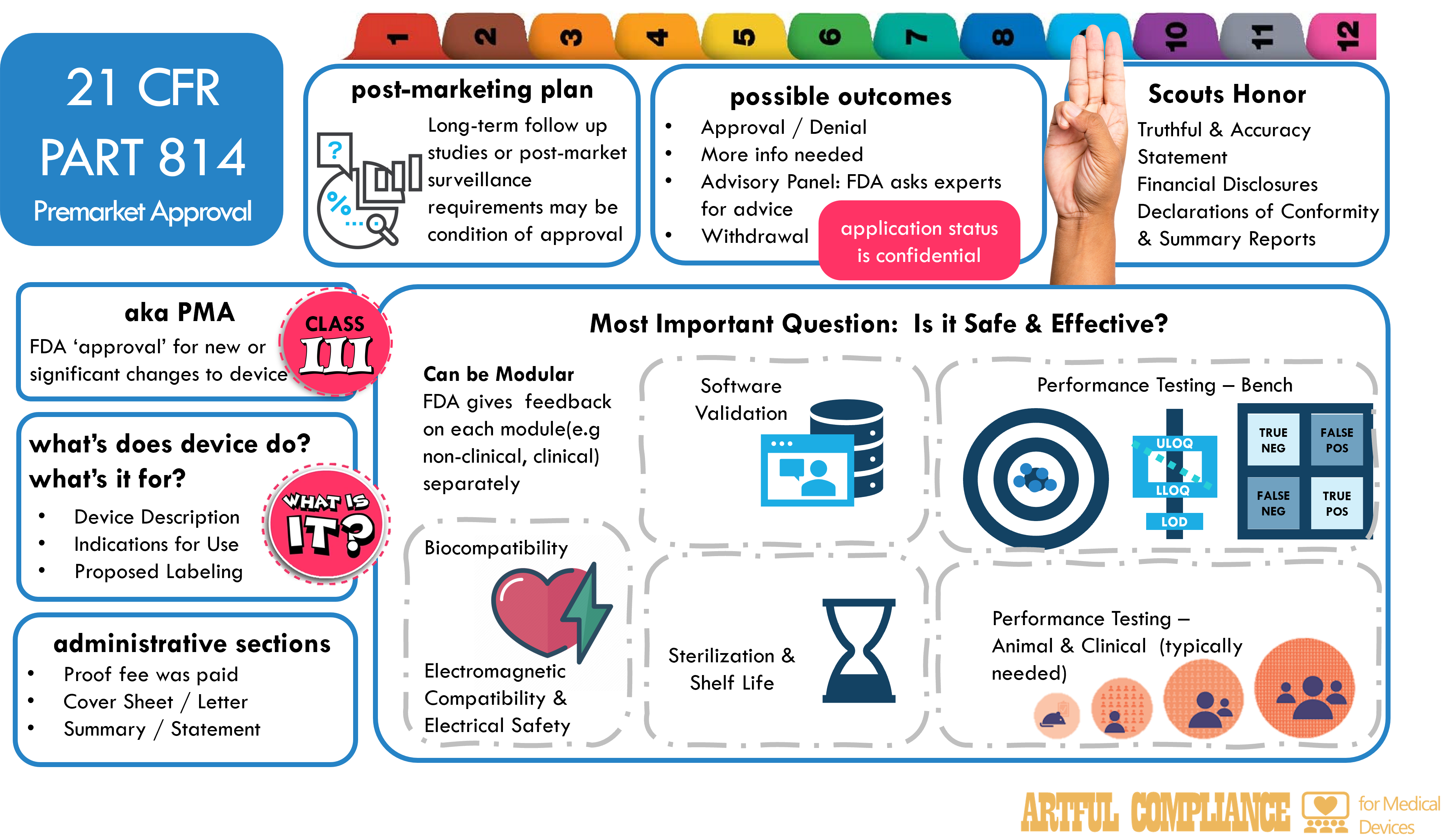
The FDA needs to agree a medical device is reasonably safe and effective before you can sell it in the US. For Class III (high risk devices), this agreement is called ‘approval’. To get ‘approval’, you’ll need to submit evidence your device is safe and effective (a process called a ‘premarket approval’). In this article, we will explain what a PMA submission is, when it is required, how to prepare it, and what to expect from the FDA review process.
What is a PMA submission?
A PMA submission is a document provides evidence to the FDA of reasonable assurance of safety and effectiveness of a device when used as intended. A PMA submission must include all information about the device, such as its design, manufacturing, labeling, performance testing, clinical data, and risk analysis. A PMA submission may also include a request for an advisory panel meeting, where a group of experts will review and discuss your device and make recommendations to the FDA. A PMA submission is the most rigorous and costly type of premarket submission for medical devices.
When is a PMA submission required?
You need to submit a PMA to the FDA if:
- Your device is high-risk, or does not qualify for a 510K or de novo. See my article on 21 CFR Part 807-E which covers pre-market notification (510Ks) for moderate-risk devices, which is a less rigorous and costly type of submission for devices that are substantially equivalent to an existing device.
You do not need to submit a PMA if:
- Your device is exempt by regulation or order (see FDA’s list of exempt devices)
- Your device is for export only
- Your device is for investigational use only (see 21 CFR Part 812).
What information is required in a PMA submission?
A PMA submission must include the following information:
What Does Device Do? What is it for?
- Device description: A description that provides the name, the classification, the product code, the regulatory status, the marketing history, and the physical and technical characteristics of the device
- Indications for use statement: A statement that describes the intended use of the device, including the patient population, the disease or condition, and the mode of application
- Proposed labeling: A copy of the proposed labeling for the device, including the instructions for use, the indications, the contraindications, the warnings, the precautions, and the adverse effects
Answer Most Important Question: Is it Safe and Effective?
- Performance testing: A description of the performance testing (bench, animal, and/or clinical) and results for the device, to demonstrate its safety and effectiveness for its intended use.
- Animal and clinical testing: A description of any animal and clinical testing and results for the device, including the study design, the study population, the study endpoints, the statistical analysis, and the ethical considerations. Clinical studies are typically required for a PMA.
- Sterilization and shelf life: A description of the sterilization method, the sterility assurance level, the packaging, the storage conditions, and the shelf life of the device, if applicable
- Biocompatibility: A description of the biocompatibility testing and results for the device, if it contacts human tissues or fluids
- Software: A description of the software design, development, verification, validation, and hazard analysis for the device, if it contains software
- Electromagnetic compatibility and electrical safety: A description of the electromagnetic compatibility and electrical safety testing and results for the device, if it emits or is affected by electromagnetic radiation.
- Additional information: Any other information that may be relevant or requested by the FDA, such as risk analysis, literature review, reference standards, etc.
Honesty Stuff
- Truthful and accuracy statement: A statement that certifies that all information in the submission is truthful and accurate, and that no material fact has been omitted
- Financial certification or disclosure statement: A certification or a disclosure that are required for clinical investigators who conducted or participated in clinical studies that support the PMA (see 21 CFR Part 54)
- Environmental impact statement, or a claim for categorical exclusion
Administrative Stuff
- User fee cover sheet: A form that identifies the submitter and the type of submission, and confirms the payment of the FDA’s fee.
- PMA cover letter: A letter that summarizes the purpose and content of the submission, and provides the contact information of the official correspondent.
- PMA summary or statement: A summary that provides an overview of the device, the predicate device, and the substantial equivalence comparison, or a statement that agrees to make the complete PMA available upon request.
How to format a PMA submission?
A PMA submission are now required to be electronic and submitted using the FDA’s Electronic Submissions Gateway (ESG) and eCopy Program. A PMA submission can also be formatted in different ways depending on the type and number of devices. For example:
- Multiple devices: A PMA can include more than one device, if they have the same intended use, the same technological characteristics, and the same performance specifications. The submission should clearly identify and distinguish each device and its corresponding information.
- Modular PMA: A PMA can be submitted in sections or modules, such as device description, preclinical data, clinical data, etc. These can be submitted over time, rather than all at once. THe DA will review each module separately and provide feedback before the next moduleis submitted. The submission should follow the FDA’s Modular PMA Program Guidance and include a table of contents for each module.
- Expedited PMA: A PMA can be eligible for expedited review, if the device is intended to treat or diagnose a life-threatening or irreversibly debilitating disease or condition, and meets certain criteria. The submission should follow the FDA’s Expedited Access Pathway Program Guidance and include a data development plan.
What is a PMA summary?
A PMA summary sometimes called a ‘technical summary’, is a document that provides an overview of the device, the predicate device, and the substantial equivalence comparison. It is intended to be a concise and accurate summary of the PMA submission, and it is made available to the public by the FDA. A PMA summary must include the following information:
- Applicant’s and Correspondent’s name and address: The name and address of the person or entity who submitted the PMA and who is authorized to communicate with the FDA about the PMA
- Device name: The trade or proprietary name, the common or usual name, and the classification name and product code of the device
- Device description: A brief description of the device, including its function, principle of operation, and technological characteristics
- Intended use: A statement of the intended use of the device, including the patient population, the disease or condition, and the mode of application
- Summary of technological characteristics: A summary of the technological characteristics of the device and the predicate device, and a comparison of their similarities and differences
- Summary of performance data: A summary of the performance data (bench, animal, and/or clinical) that support the safety and effectiveness of the device for its intended use
- Conclusions: A statement of the conclusions drawn from the comparison of the device and the predicate device, and the demonstration of substantial equivalence
What is a PMA statement?
A PMA statement is a document that agrees to make the complete PMA submission available to any person within 30 days of a written request. It is an alternative to a PMA summary, available under certain conditions, and it is not made available to the public by the FDA. A PMA statement must include the following information:
- Statement that all information in the submission is truthful and accurate, and that no material fact has been omitted.
- Statement that the submitter will make the complete PMA available to any person within 30 days of request and specifies the address where such requests should be sent
- Name, title, address, telephone number, and signature of the official correspondent with the FDA
What to expect from the FDA review process
- Submission acceptance: The FDA will check if the submission is complete and meets the minimum requirements for review. This usually takes 15 days once received. The FDA will issue an acceptance or a refusal letter to the submitter.
- Substantive review: The FDA will conduct a detailed review of the submission to determine if the device is safe and effective for its intended use, and that it provides a reasonable assurance of safety and effectiveness. This usually takes 180-320 days from the date of acceptance. The FDA will communicate with the submitter during the review to request additional information, clarify issues, or resolve problems.
- Advisory Panel: The FDA may also convene an advisory panel meeting to obtain expert opinions and recommendations on the device. The FDA will issue an approval or a denial order to the submitter.
- Marketing approval: If the FDA approves the device, the submitter can market the device in the U.S. subject to the general and special controls applicable to the device. The FDA will list the device on its PMA database and publish the PMA summary or statement on its website.
Recent Comments