Investigating A Medical Device
Consider this Catch-22:
- You can’t distribute a medical device before getting FDA approval.
- You can’t get FDA approval without evidence the medical device works.
- You can’t get evidence a device works without using the medical device.
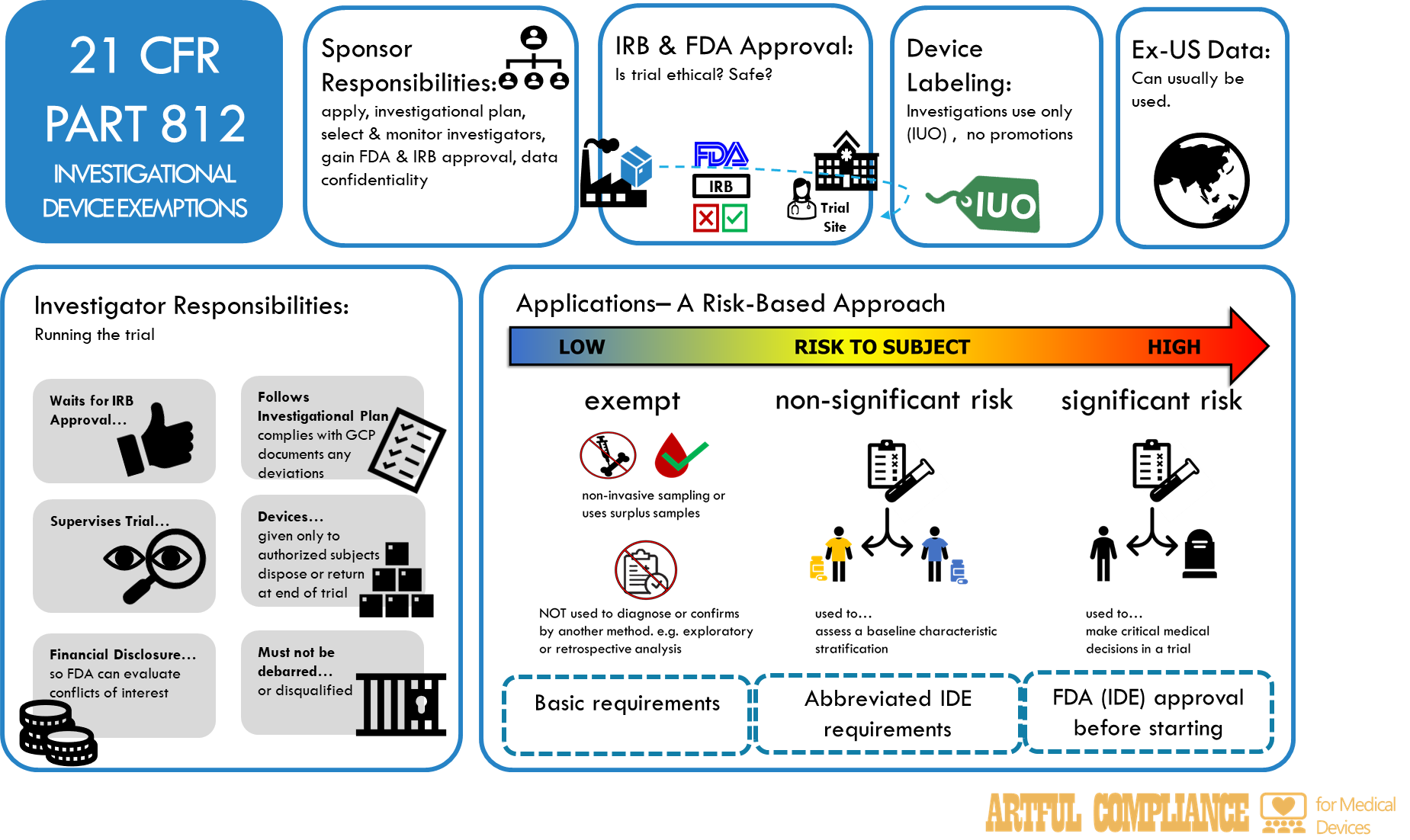
The FDA solved this problem with 21 CFR Part 812, which allows an exemption to distribute a device for a clinical trial to investigate it (i.e. ‘investigational device exemption’ or ‘IDE’).
21 CFR Part 812 takes a risk-based approach to IDEs, as well as the procedures for obtaining IRB and FDA approval, and the responsibilities of sponsors and investigators who conduct device studies. It also addresses how investigational devices must be labeled.
Risk-Categories of Investigational Devices
The FDA classifies investigational devices into three categories based on the level of risk they pose to human subjects:
- exempt,
- non-significant risk (NSR), and
- significant risk (SR).
The risk category determines the type and extent of regulatory oversight required for the device study.
Exempt devices are those that do not require an IDE from the FDA, because they meet one of the following criteria:
- They are legally marketed devices that are used in accordance with their approved labeling
- They are diagnostic devices with little or no risk to subjects (see 21 CFR 809.10(c))
- Testing is not to determine safety or effectiveness of device, and does not put subjects at risk
- They are custom devices not intended for general distribution.
Significant Risk (SR) devices are those that:
- Are implants
- Support or sustain human life
- Are of substantial importance to diagnose, cure, mitigate, or treat disease, or otherwise prevent impairment of human health
- Present a potential for serious risk to the health, safety, or welfare of a subject
Non-Significant Risk (NSR) devices are everything else.
Actions Required Based on Risk
Exempt devices
The sponsor does not need to submit an IDE application to the FDA, but must:
- Obtain approval from an IRB before initiating the study
- Label the device as investigational
- Obtain informed consent from subjects
NSR devices
The sponsor does not need to submit an IDE application to the FDA, but must. The sponsor must do everything required for an exempt device AND also comply with the abbreviated requirements in 21 CFR 812.2(b), which include:
- Conduct the study per the approved protocol
- Monitor the study and ensure that any unanticipated adverse device effects are reported to the FDA and the IRB
- Maintain certain records and reports (see 21 CFR 812.140(b)(4) and (5))
SR devices
The sponsor must submit an IDE application to the FDA and obtain approval from both the FDA and an IRB before initiating the study. The sponsor must also comply with the full requirements in 21 CFR Part 812, which include everything for exempt and NSR devices AND:
- Conduct the study in per any conditions of approval imposed by the FDA or the IRB
- Maintaining all records and reports as required by 21 CFR Part 812. NOTE: There are considerably more records and reports needed for SR devices.
IRB approval process for device studies
The Institutional Review Board (IRB) is an ethics committee responsible for reviewing and approving human studies to ensure that they are ethically sound and protect the rights and welfare of human subjects. The IRB must follow the general requirements in 21 CFR Part 56 (IRB), as well as the specific requirements in 21 CFR Part 812 for device studies.
The IRB must review and approve:
- The research protocol
- The informed consent document
- The investigational device brochure (if any)
- The investigator’s qualifications
- The site where the study will be conducted
- Any other relevant information
The IRB must also determine whether the device is NSR or SR based on the information provided by the sponsor. If the IRB disagrees with the sponsor’s initial NSR determination, it must notify the sponsor and the FDA within five working days.
FDA approval process for device studies
The FDA is responsible for reviewing and approving IDE applications for SR devices to ensure that they are scientifically valid and protect public health. The FDA will follow the general requirements in 21 CFR Part 50 (Protection of Human Subjects), as well as the specific requirements in 21 CFR Part 812 for IDE applications.
The FDA will review and approve:
- The research protocol
- The risk analysis
- The device description
- The manufacturing information
- The preclinical and clinical data (if any)
- The investigator’s qualifications
- The informed consent document
- The investigational device brochure (if any)
- Any other relevant information
The FDA will also determine whether the device is SR or NSR based on the information provided by the sponsor. If the FDA disagrees with the sponsor’s initial SR determination, it must notify the sponsor and the IRB within 30 days.
Sponsor Responsibilities
The sponsor is the person or entity who initiates a device study, or who assumes responsibility for a study initiated by another person or entity. The sponsor is typically the manufacturer of the device, a clinical investigator, an academic institution, or a contract research organization.
The sponsor must:
- Obtain an IDE from the FDA (if required) and an approval from an IRB before initiating a device study
- Select qualified investigators and provide them with the necessary information and training to conduct the study
- Provide the investigators with the investigational devices and ensure that they are properly labeled, stored, distributed, and accounted for
- Obtain agreements from the investigators to comply with the protocol, the IDE regulations, and any other applicable regulations
- Monitor the progress and conduct of the study and ensure that any problems or deviations are corrected or reported
- Report any unanticipated adverse device effects, withdrawals of IRB or FDA approval, progress reports, final reports, and any other information as required by 21 CFR Part 812
- Maintain records and reports as required by 21 CFR Part 812
Investigator responsibilities
The investigator is the person who actually conducts a device study, or who supervises the conduct of a device study by other persons under his or her direction. The investigator may be a physician, a nurse, a technician, or any other person qualified by training and experience to conduct the study.
The investigator must:
- Obtain approval from an IRB before initiating a device study
- Obtain informed consent from each subject before enrolling them in the study
- Conduct the study per the approved protocol and any conditions of approval imposed by the IRB or the FDA
- Use the investigational device only for the purposes and in the manner specified in the protocol
- Control the investigational device and ensuring that it is not used for any other purpose or by any other person
- Report any unanticipated adverse device effects, deviations from the protocol, changes in the study status, and any other information as required by 21 CFR Part 812
- Maintain records and reports as required by 21 CFR Part 812
Device labeling requirements for investigational devices
The labeling of investigational devices is regulated by 21 CFR Part 812 to ensure that they are clearly identified as such and that they provide adequate information for their safe and effective use.
The labeling of investigational devices must:
- Bear the statement “CAUTION — Investigational device. Limited by Federal (or United States) law to investigational use.”
- Identify the device, the name and address of the manufacturer or sponsor, and any relevant information such as expiration date, lot number, serial number, etc.
- Provide directions for use, warnings, precautions, potential adverse effects, contraindications, and any other information necessary for the protection of subjects
- Not contain any false or misleading statements or claims about the safety or effectiveness of the device.
Recent Comments