Recalls are ‘Voluntary’… …Until They Are Not
21 CFR Part 810 is one of the more serious and impactful regulations for most medical device manufacturers to understand, as it can impact a company’s reputation and bottom line. But for Laboratories worried about the impact of Laboratory Developed Tests (LDTs) becoming IVDs, it’s unlikely to cause concern, because the IVD is ‘distributed’ to only one customer (who is also the manufacturer), which majorly simplifies the actions needed to comply.
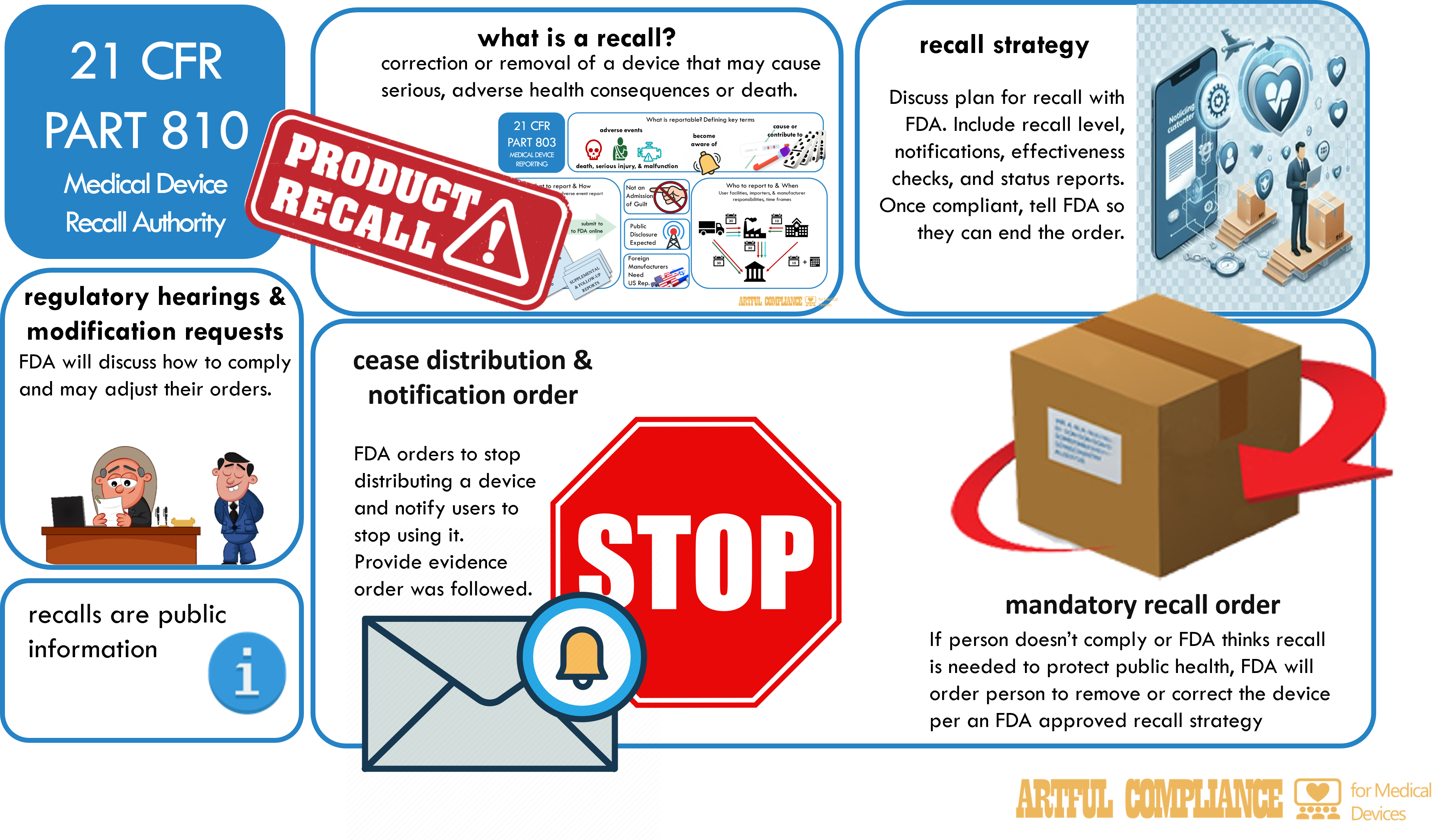
In my article about 21 CFR Part 806 Corrections and Removals: The Recall Regulation, I discussed what a recall is, that you must report it to the FDA, and how. Here’s a quick summary.
A recall is a voluntary action taken by a manufacturer, importer, or distributor to correct or remove a device that is in violation of the FD&C Act or that may present a risk to health. It may include if you:
- Repair, modify, adjust, relabel, or destroy the device
- Inspect the device even if you don’t physically remove it from the customer.
- Monitor patients due to suspected device issues.
- Notify the device users of the problem and provide instructions on how to handle or dispose of the device
Recalls occur when:
- the FDA determines there is a reasonable probability that a device intended for human use would cause serious adverse health consequences or death and requests a recall.
- you start a correction or removal of a device to reduce a risk to health.
Voluntary, in this context is a relative term. The regulation doesn’t mean recalls are optional. It just means that are voluntary relative to other options, for instance if the FDA decides to enforce its medical device recall authority under the powerful Federal Food, Drug, and Cosmetic Act (FD&C Act), as we will describe in this article.
Cease Distribution and Notification Order
The word ‘order‘ sounds serious, doesn’t it? It’s worth remembering at this point that Federal Agencies like the FDA have actually legal power, that was passed to them by the US Congress. They can, and do, issues ‘orders’ much in the way a police officer can. And like a police officer, they have the very real ability to physically walk into a company and shut down activities they find illegal.
A cease distribution and notification order (CDANO) is the equivalent of a police officer give a lawful order to stop doing something. It must be obeyed, or a company risks more serious consequences. This type of order issued by the FDA requires a manufacturer or importer to immediately:
- Stop distributing a device
- Notify all persons who received, purchased, or used the device of the order and the reason for it
- Instruct them to stop using the device and to take appropriate action to prevent further use
Just as a police officer issues an order when they have just cause, a cease distribution and notification order may be issued when:
- The FDA has credible evidence that the device presents an unreasonable risk of substantial harm to the public health
- The FDA has consulted with the manufacturer or importer regarding the evidence and the need for an order
- The FDA has provided an opportunity for an informal regulatory hearing on whether the order should be issued
The FDA will share the reason with you when they issue the order.
Complying With a CDANO
I remember this as “Can’t Distribute Any New Orders” because to comply with a cease distribution and notification order, the manufacturer or importer must Immediately stop distributing the device and with 24 hours confirm in writing to the FDA that distribution has ceased.
A single-site IVD would have few logistical issues notifying their single customer, though this is not to say you wouldn’t potentially still have issues in maintaining testing as the lab runs out of IVD reagent inventory. But that’s little ins relation to a large-scale manufacturer. As you can imagine, depending on the number devices out there and a large distribution chain, this can be quite the scramble. And it gets worse, you also need to provide proof of what you did.
Within 10 working days of receiving the order, you have to submit a written report to the FDA that includes:
- The name, address, telephone number, and email address of each person who received, purchased, or used the device
- The date on which each person received, purchased, or used the device
- A copy of each notification sent to each person
- A description of any actions taken by each person in response to the notification
- A description of any problems encountered in complying with the order
Recall Strategy
This is where recall strategies come into play.
A recall strategy is a planned, specific course of action to be taken by the manufacturer or importer in response to a cease distribution and notification order or a mandatory recall order. A recall strategy addresses:
- The depth of the recall (i.e., how far down the distribution chain the recall will extend)
- The need for public warnings (i.e., whether any announcements or notices should be issued to inform the public of the recall)
- The effectiveness checks (i.e., how to verify that all persons who received, purchased, or used the device have been notified and have taken appropriate action)
To develop and implement a recall strategy, the manufacturer or importer must:
- Consult with the FDA on the elements of the strategy
- Submit a written proposal of the strategy to the FDA within 15 working days of receiving a cease distribution and notification order or a mandatory recall order
- Obtain approval from the FDA before implementing the strategy
- Follow up with periodic status reports on the progress and results of the strategy
Mandatory Recall Orders – It’s Serious Now
Some people just don’t obey Officers. But then what? At this point, the manufacturer had an option to ‘voluntarily’ recall, and has been issued a cease distribution and notification order. They’ve had the chance to discuss a recall strategy with the FDA to comply. At this point most manufacturers choose to comply.
If not, a mandatory recall order can be issued by the FDA and requires a manufacturer or importer to recall a device. It may be issued when the FDA determines that the device presents an unreasonable risk of substantial harm to the public health and that the manufacturer or importer has not taken sufficient action to recall the device.
A mandatory recall order is public in nature and has serious consequences for the manufacturer or importer. The FDA will:
- Publish a notice of the order in the Federal Register and on its website
- Notify all persons who received, purchased, or used the device of the order and the reason for it
- Instruct them to stop using the device and to take appropriate action to prevent further use
- Monitor and enforce compliance with the order
- Impose civil penalties for violations of the order
It’s the last two items that make this option the most serious. The FDA is not toothless. They can revoke a manufacturer’s registration, effectively shutting down production. Still not complying? They can call in the local police and show up at your facility. They can take the product away physically, lock up the product in a room, or lock up the entire facility and send your staff home. For foreign manufacturers, they can hold the product in customs. F-around and find out.
What if You and the FDA Don’t Agree or the Situation Changes?
There is recourse if you end up at odds with the FDA. Say you don’t agree with their CDANO, or can’t agree on a recall strategy?
A regulatory hearing is an informal proceeding conducted by an FDA official who is independent of the CDANO or a mandatory recall order. It provides an opportunity for:
- The manufacturer or importer to present information and views on whether there is credible evidence that supports issuing or continuing an order
- The FDA to explain its rationale for issuing or continuing an order
A modification request is a written request submitted to the FDA to modify or terminate a cease distribution and notification order or a mandatory recall order. A modification request may be based on:
- New information or evidence that shows that the order is not warranted or is no longer necessary
- Changes in the circumstances or conditions that led to the issuance of the order
- Compliance with the terms and requirements of the order
To request a regulatory hearing or a modification request, the manufacturer or importer must:
- Submit a written request to the FDA within 10 working days of receiving a cease distribution and notification order or a mandatory recall order
- Include a statement of the factual and legal basis for the request and any supporting information or evidence
- Specify whether the request is for a regulatory hearing, a modification, or both
Recent Comments