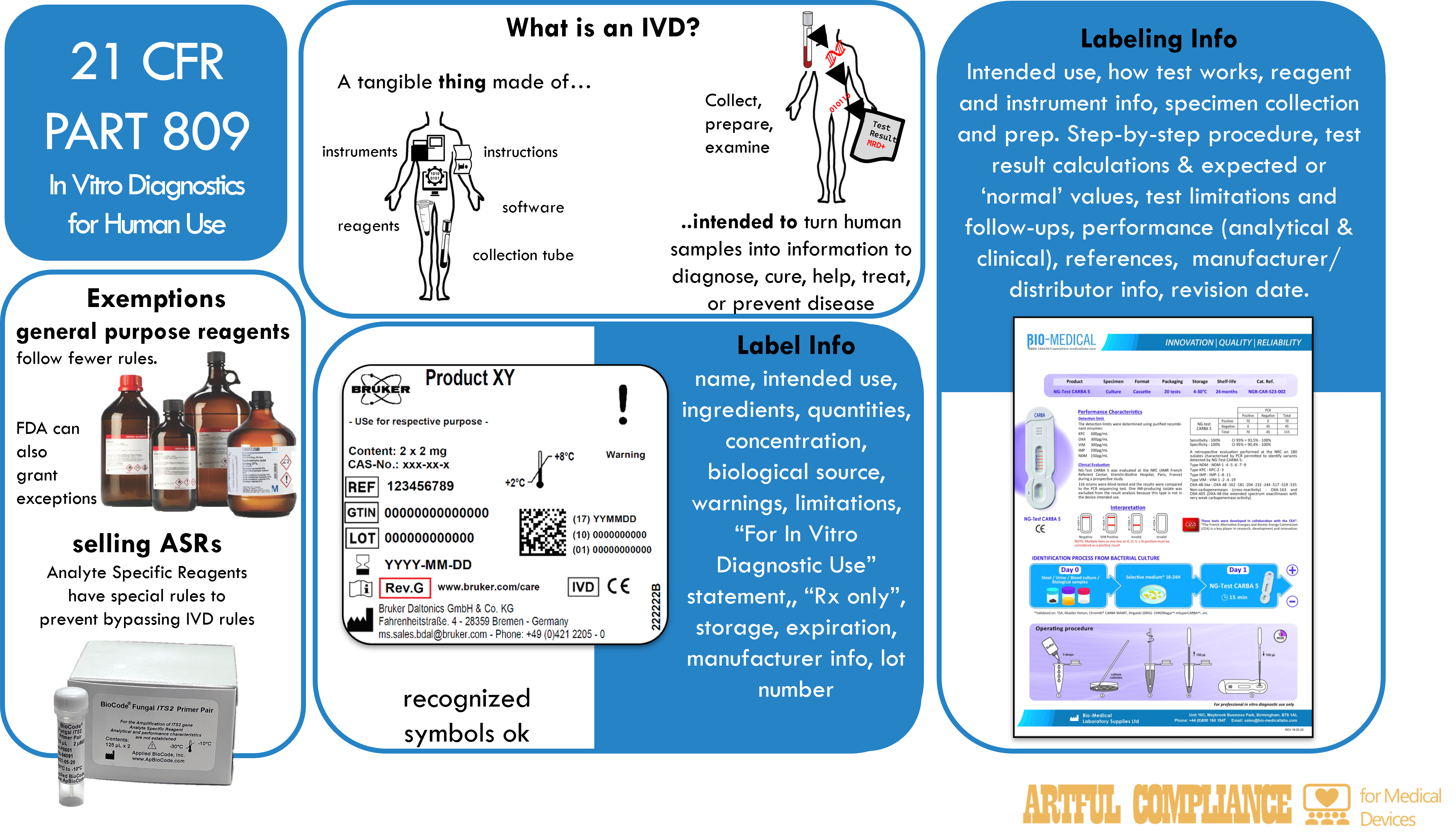
This regulation contains the labeling requirements for in vitro diagnostic products, which are devices or biological products used for testing specimens from the human body. Due to their special nature, the labeling requirements are different than the general medical device labeling requirement. The focus of the regulation is the information that must be stated on the label and the accompanying labeling, but there are also rules about who certain IVDs can be sold to that attempt to put limitations on the use of Laboratory Developed Tests.
Part A. Definition of an IVD
The regulation begins by defining what an IVD is,
reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
Part B. Requirements for label and labelling of an IVD (809.10)
Labels vs Labelling
If you mistake the two, it will cause confusion and chaos in understanding the regulations.
- Labels are the written, printed, or graphic matter on the immediate container (occasionally an outer container if there is a space constraint) of a medical device. Labels include tags, stickers, or other attachments to the product or its container.
- Labeling is a broader term– it includes all labels, but also the package insert, which is a document that provides instruction for use and other information necessary for the user to fully understand the product, its use, and limitations.
Labels On the Product
The label for an in vitro diagnostic product must state the label info (see list below), and other important information as applicable. The label must be legible and appear on the immediate container unless the container is too small, in which case it can go on outer containers or wrappers. The purpose of the lot number is to allow tracing to full manufacturing history, including all sub-parts of a kit or instrument.
Label information:
(1) product name, (2) intended use, (3) reagents name ingredients, (3) quantity of the product or tests, (3) concentration of reactive ingredients, (3) biological source, (3) biological activity,(4) warnings, (4) the statement “For In Vitro Diagnostic Use”, (4) any other limitations, (4) for prescription products: “Rx only” or “Caution: Federal law restricts this device to sale by or on the order of a ___”, (e.g. “physician”), (5) storage instructions, (5) reconstitution instructions, (6) expiration date (or other indicators), (7) quantities individual reagents, (8) manufacturer info, (9) lot number, and other information as applicable.
Labeling Accompanying the Product
The labeling that comes with the product, such as a package insert, must state the information below, in order.
Labeling information:
(1) name, (2) intended use, (2) type of procedure, e.g., qualitative or quantitative, (3) Summary and explanation of the test, (4) chemical, physical, physiological, or biological principles of the procedure, (5) Reagent information, (6) Instrument information, (7) specimen collection and preparation information, (8) Procedure ( step-by-step, and any points that may be useful in improving precision and accuracy), (9) description of result and how to calculate them, (10) Limitation of the procedure, (10) results that indicate further testing is needed, (11) expected values (e.g. range), (11) how the range(s) was established (e.g. ‘normal’ population), (12) specific performance characteristics, (13) Bibliography and references, (14) manufacturer/packer/distributor info, (15) revision date.
This information is often lengthy, and as a result rather than printing each time, manufacturers provide a document that shares where the information is available on a website.
Exceptions or alternatives
The FDA can grant exceptions or alternative to any labeling requirement if compliance with the requirement could adversely affect the safety, effectiveness, or availability of the products. The request for an exception or alternative must be submitted in writing and include rationale.
General purpose laboratory reagents and equipment
Labels of general purpose laboratory reagents and equipment that are not intended for a specific diagnostic application are exempted if their labeling meets a small set of requirements: description, composition, purity, quality, warnings, storage instructions, quantity, and lot number of the product.
Analyte specific reagents (ASR)
The labels of analyte specific reagents (e.g. monoclonal antibodies, DNA probes, viral antigens, and ligands) is exempted if their labeling meets a small set of requirements: for example name, quantity, purity, quality, warnings, storage instructions, expiration date, manufacturer, lot number, and the statement “Analyte Specific Reagent. Analytical and performance characteristics are not established” or “Analyte Specific Reagent. Except as a component of the approved/cleared test (Name of approved/cleared test), analytical and performance characteristics are not established”.
ASR labeling must not make any statement regarding analytical or clinical performance. The laboratory that develops an in-house test using the analyte specific reagent must inform the ordering person of the test result by appending the statement “This test was developed and its performance characteristics determined by (Laboratory Name). It has not been cleared or approved by the U.S. Food and Drug Administration.”
Use of symbols
The labeling of in vitro diagnostic products may use symbols in place of text if
- the symbol is accompanied by explanatory text adjacent to the symbol, or
- the symbol is contained in a standard recognized by the FDA, or
- the symbol is established by a standards development organization and is determined by the manufacturer to be likely to be read and understood by the ordinary individual.
The symbol must be used according to the specifications of the standard and must be explained in a symbols glossary that is included in the labeling.
Part C—Requirements for Manufacturers and Producers
IVDs are Medical Devices
This part is about selling IVDs. FDA reminds us that IVDs are medical devices, and sometimes biological products. As such, manufacturers of IVD products must comply with good manufacturing practices and, if applicable, biological product standards.
Selling Analyte Specific Reagents
There are also specific rules about selling analyte specific reagents (ASR’s). The general idea is that the FDA wanted to prevent ASR manufacturers from circumventing the 510K or PMA process by selling ASRs as RUO reagents to laboratories ad telling the labs they could use them as Laboratory Developed Tests. As a result, ASRs must be labeled and advertised according to specific requirements. Laboratories that use ASR’s to develop in-house tests must inform the ordering person of the test result and the test status.
Over-the-counter test sample collection systems for drug abuse testing
There are special rules for labeling of over-the-counter (OTC) test sample collection systems for drugs of abuse testing. These are restricted devices that must ensure sample testing is performed in a laboratory using approved or recognized screening tests. The labeling of these devices must provide adequate instructions and information for the lay users.
Recent Comments