What is a Recall?
‘Recall’ of a medical device is when a manufacture takes action to address a problem with a device that violates FDA law OR has a defect that could be a risk to patient healt.. The action may be to repair, modify, adjust, relabel, inspect, or destroy it. They come in two flavors depending on where the action happens 1) Correction and 2) Removal.
‘Correction’ is when the action to address the problem occurs without physically removing it from where the device is used or sold. It’s not always feasible or necessary to take back a device to fix an issue. A device might simply need to be checked or adjusted by a technician, or new software installed. Sometimes a correction is just to provide new instructions for use.
‘Removal’ is when the action to address the problem is to physically remove it from where it is used or sold.
History of the Recall Regulation
Before the Safe Medical Devices Act of 1990 (SMDA), reporting recalls of medical devices was voluntary. It was like letting students decide which exams to turn in—the teachers are unlikely to get an accurate picture of student performance or know how to help failing students. Congress believed there was under-reporting of recalls to the FDA. In turn, this left the FDA unable to do its job of evaluating device-related problems and how those problems were being corrected.
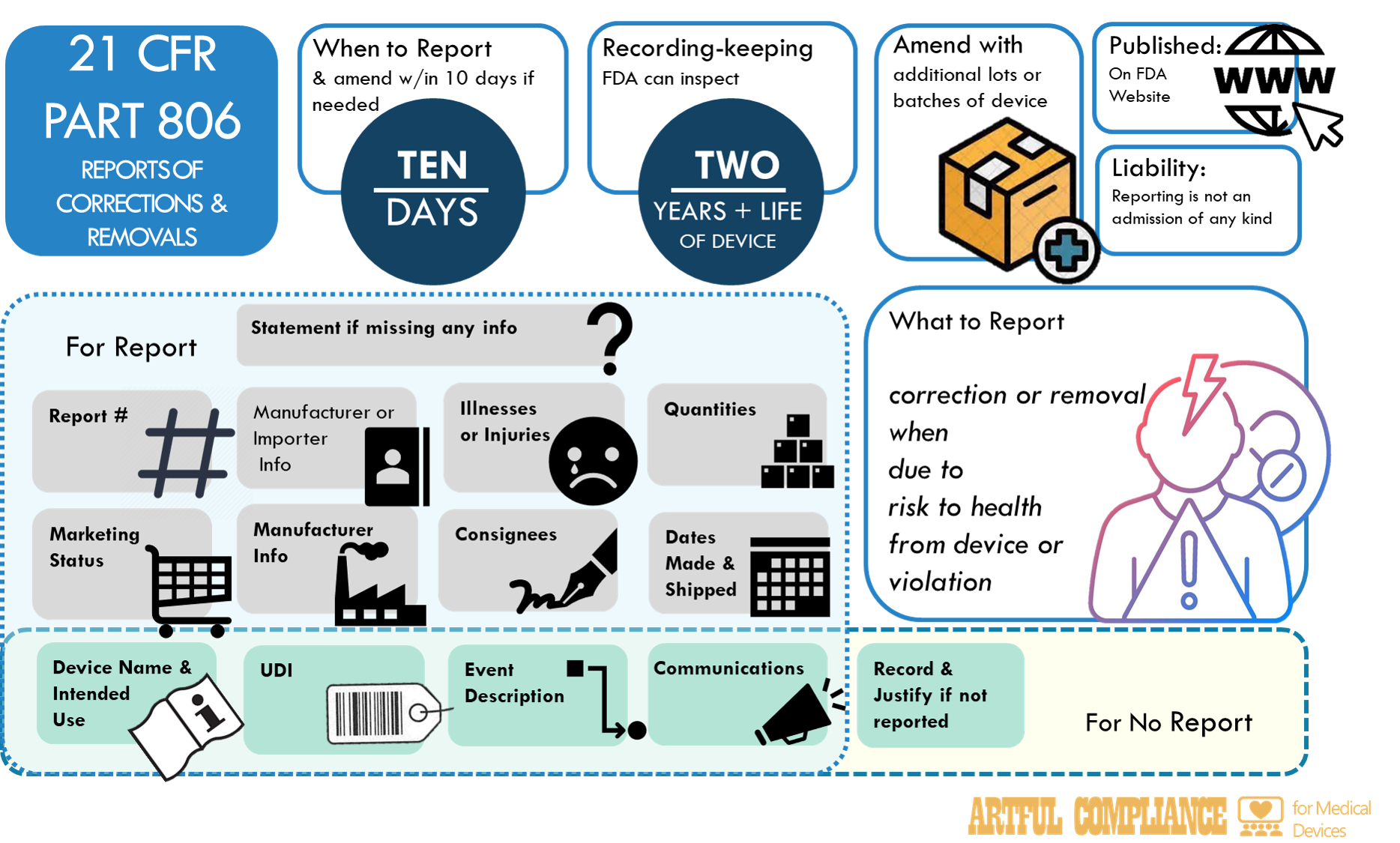
After the SMDA, the FDA created 21 CFR Part 806, Reports of Corrections and Removals. It mandated manufacturers (including Ex-US) to promptly report certain types of device corrections and removals to the FDA. Manufacturers also had to maintain records of all corrections and removals regardless of whether they met the criteria for reporting to the FDA. 21 CFR Part 806 is a scant 4 pages long, and not a challenging read, but here’s the top things you should know.
Not Everything Needs to Be Reported
There are some recall actions you don’t need to report (21 CFR 806.1(b)):
| Improvements not related to health risks or violations | Actions that improve performance or quality of a device but do not reduce a risk to health posed by the device or remedy a violation of device laws. (see below for definition of ‘risk to health’) |
| Market withdrawal | Actions that involve a minor violation of the act that would not be subject to legal action by FDA or that involves no violation of device laws. |
| Routine servicing | Any regularly scheduled maintenance of a device (e.g. planned part replacement, calibration, battery replacement), and responses to normal wear and tear. Unexpected repairs or replacements or unexpected frequency of this activity are not routine servicing. |
| Stock recovery or rotation | If a device has not been marketed or has not left the direct control of the manufacturer, i.e., is located on the premises owned, or under the control of, the manufacturer, and no portion of the lot, model, code, or other relevant unit involved in the recall been released for sale or use. |
| Actions already reported as part of Adverse Event | If you’ve already reported the recall in following the adverse event reporting (see 21 CFR Part 803) process, you don’t need to do it again. |
Of particular interest to LDT laboratories will be the stock recovery exemption. It means that even if a lot of reagents has been released to the laboratory, so long as the laboratory can show the lot has not been used on patient samples, it can be argued that it can be removed from the lab without considering it a recall.
Recalls to Reduce A Risk To Health Must be Reported
A lot of the decision to report or not hinges on the words ‘risk to health’, so let’s explore that a little.
Risk to health means (1) A reasonable probability that use of, or exposure to, the product will cause serious adverse health consequences or death; or(2) That use of, or exposure to, the product may cause temporary or medically reversible adverse health consequences, or an outcome where the probability of serious adverse health consequences is remote
Medical device manufactures of typical IVDs know risk management pretty well— it is baked into ISO 13485, and ISO 14971. But laboratory professionals responsible for LDTs may not be as familiar with definitions of risk. In simple terms, “risk” considers probability (likelihood) and severity (consequences). The FDA defines three ‘classes’ (I, II, III) due to use of a device that constitute a reportable risk to health:
| Situation | Probability | Severity |
| Class I | Reasonable | Serious adverse heath consequences or death |
| Class II | Possible (“may”) | Temporary or medically reversible |
| Class II | Remote | serious adverse health consequences |
| Class III (voluntary to report) | Not likely | adverse health consequences |
This means that to determine if there is a ‘risk to health’ your team must first estimate (using available data) both the probability and severity of problem with the IVD, and depending on that evaluation determine if the problem meets the criteria in this table. If you aren’t sure, you should report, and keep in mind that the FDA will makes its own evaluation (aka a ‘health hazard assessment” to classify your recall.
Report Promptly
The FDA didn’t leave what this means up to the manufacturer. Promptly means within ten working days of starting the recall.
How to Report
The FDA doesn’t have a form for reporting recalls, but requires manufactures to include all of the following (21 CFR 806.10(c)):
- Report number: registration, date, sequence, and type
- Manufacturer or importer: representative info
- Device name and use
- Device marketing status: number
- Device ID: UPC, model, catalog, code, lot, or serial number
- Manufacturer info: if different from reporter
- Event and action: description
- Illness or injuries: report numbers if any
- Number of devices and batches: for correction or removal
- Manufacture or distribution date: device life
- Consignees info: distribution details
- Communications copy: recipients not in element
- Reason and date: for missing information
Moreover, if you end up extending recall to other products, you have to ‘promptly follow up with another report amending the original report. You have two ways to get this info to the FDA
- eSubmitter: Electronic Submission of 806 Reports of Corrections and Removals
- E-mail the FDA ORA Recall Coordinator: ORA Recall Coordinators | FDA
A Report is Not a Statement of Guilt
As with adverse event reporting, reporting a recall does not mean the manufacturer is admitting their device caused illness or injury. The report is legally protected from being used in that way.
Record all Recalls, Even the Non-Reported Ones
Even if the recall actions don’t need to be reported, you must maintain records of those actions. The list of things to record is less than for reporting and includes:
- Brand name, classification, name and product code if known, and the intended use of the device.
- Identification. Unique device identifier (UDI) of the device, or other unique identifier code and model/catalog #, and lot or serial # if known.
- Description of the event(s) giving rise to the information reported and the corrective or removal action that has been, and is expected to be taken.
- Justification for not reporting the correction or removal action to FDA, including conclusions and any followups, and be reviewed and evaluated by a designated person.
- Copy of all communications regarding the correction or removal.
The FDA is allowed to (and will likely) inspect these records in the future.
Recall Reports are Publicly Available
The FDA will delete confidential or trade secret information, personnel names, and medical information of patients, then publish the reports on: Recalls, Market Withdrawals, & Safety Alerts | FDA
OK, What next?
A recall is a voluntary action that can be taken by a manufacturer, or at the FDA’s request. It’s almost always a better choice than seizure or other court actions. Make no mistake– the FDA can and will use force if a a manufacturer does not voluntary recall devices to their satisfaction. The process by which the FDA evaluates reported recalls, classifies recalls, asks for or mandates a recall, or decides whether the manufacturer’s recall plan is good enough is described in 21 CFR 7. In also discuss the more police-like actions the FDA can take. This will be the topic of another blog.
Sources:
(1) Recalls, Corrections and Removals (Devices) | FDA. https://www.fda.gov/medical-devices/postmarket-requirements-devices/recalls-corrections-and-removals-devices. (2) FDA’s Role in Drug Recalls | FDA – U.S. Food and Drug Administration. https://www.fda.gov/drugs/drug-recalls/fdas-role-drug-recalls. (3) FDA – Recall Types – FindLaw. https://www.findlaw.com/injury/product-liability/fda-recall-types.html. (4) Recalls Background and Definitions | FDA. https://www.fda.gov/safety/industry-guidance-recalls/recalls-background-and-definitions.
Recent Comments