With the latest FDA proposed rule-making for IVDs, laboratory professionals supporting Laboratory Developed Tests (LDTs) may be asking, what does this mean for my LDT test?
In short—the FDA wants to regulate LDTs under the same rules as in vitro diagnostic (IVD) medical devices. Those rules are spelled out in 21 CFR Chapter I, Subchapter H – Medical Devices. If you are not a Regulatory Affairs or Quality Assurance professional in the traditional medical device industry (and maybe even if you are), even looking at the table of contents may be a little daunting.
The aim of this post is to make it less so. I do this by describing each part in simple terms, and giving the relevance to a LDT, should LDTs start being regulated as IVDs. Once you have the high-level overview, you can dive down into each part. I plan to release more detailed information of each part in future blogs, so stay tuned. Note, I will omit information not applicable to IVDs to cut this information down.

Part 800: General
Administrative rules for how FDA will prevent selling or using devices they deem unsafe, and how to appeal those decisions.
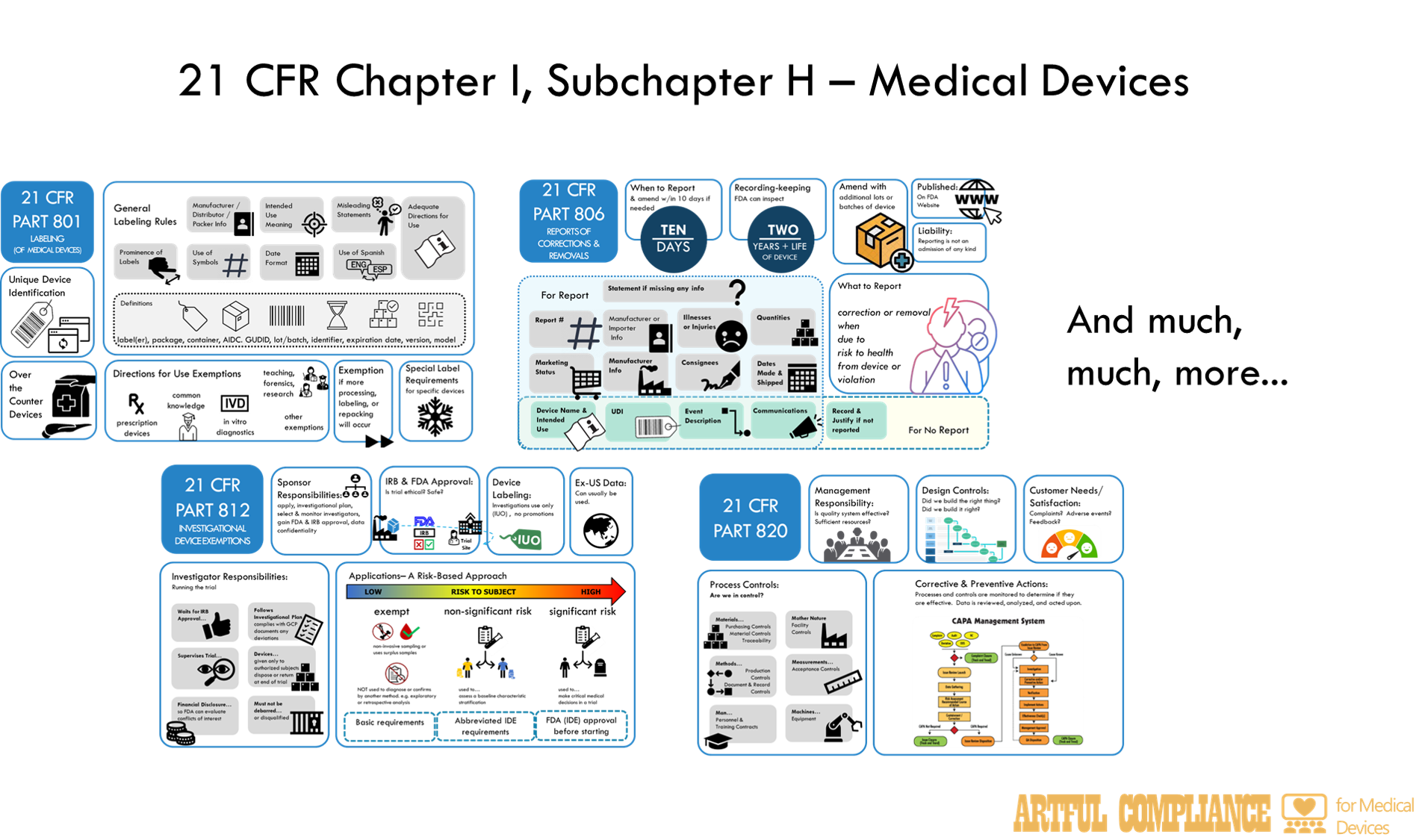
Part 801: Labeling

Rules for ‘Labeling’ which includes far more than just physical labels on IVD reagents, but also instructions for the user of reagents, instruments, or software, plus any information provided to customers about what the IVD could or should be used for. This also includes promotional material or information provided in the patient result. Note that Part 809 provides more specific rules for labeling of IVDs.
Part 803: Medical Device Reporting

Rules for reporting adverse events to the FDA and states how these reports may be shared with the public. Adverse events include, but are not limited to, patient harm due to incorrect results as a result of IVD malfunctions, or faulty user instructions or labelling.
Part 806: Medical Devices; Reports of Corrections and Removals

Rule for reporting of corrections and removals of IVDs to the FDA. This is when a lab releases IVD reagents, instruments, or software to the lab to use in testing, and then later realizes there is something ‘wrong’ with them and needs to do something to ‘fix’ the problem or take the faulty parts of the IVD away from lab users.
Part 807 (A-D): Establishment Registrations & Device Listing

Rules for how and when to register the facilities (i.e. ‘establishment’) that designs and makes the IVD, and providing a list of IVDs (i.e. ‘listing’) it makes at each facility, to the FDA. This is public information and must be updated annually.
Part 807 (E): Establishment Registrations & Device Listing

Rules for premarket notification submissions (aka ‘510k’) by which the FDA can agree (i.e. ‘clear’) that a medium risk IVD is like (i.e. ‘substantially equivalent’) to an IVD already being sold. After looking at analytical and (sometimes clinical) validation data, they can agree it is safe and effective enough to be sold.
Part 808: Exemptions From Federal Preemption of State and Local Medical Device Requirements

Federal preemption means that FDA rules can override state rules if they conflict with each other. This rule gives States a path to have different rules if they can show their rules are stricter than the FDA’s.
Part 809: In Vitro Diagnostic Products for Human Use

Specific labeling rules for IVDs including call outs for; how to label general and analyte-specific reagents, labeling instruments, what needs to go into IVD lab procedures and over-the-counter instructions, and how to describe IVD performance characteristics (e.g. accuracy, precision).
Part 810: Medical Device Recall Authority

How the FDA has the authority to issue a recall or cease distribution order. This means they can make IVD manufacturers ask users to send purchased IVD materials back at the manufacture’s expense, or stop manufactures from providing IVD products.
Part 812: Investigational Device Exemptions

If the FDA has not cleared or approved an IVD, it can still be used in a clinical trial, but is considered an ‘investigational device’ and requires an ‘exemption’ to use it. The Sponsor and manufacturer also need to follow certain rules depending on risk involved in using the IVD in the trial. For the highest risk category, a formal application to the FDA is needed.
Part 814: Premarket Approval of Medical Devices

Rules for premarket approval submissions (aka ‘PMA’) by which the FDA can agree (i.e. ‘approve’) that a high risk IVD is safe and effective enough to be sold. Both analytical and clinical validation data is generally needed, and there may be commitments to monitor the IVD or perform additional studies even after approval (i.e. ‘postapproval requirements’).
Rules for humanitarian IVDs for rare use cases (e.g. rare diseases).
This part explains how to get approval from the FDA for a new medical device that is not substantially equivalent to an existing one. It also describes the types of clinical studies that are required to demonstrate the safety and effectiveness of the device1.
Part 820: Quality System Regulations

Rules for the quality system of IVD manufacturers, which includes procedures for design, production, packaging, labeling, storage, installation, and servicing of their devices. It requires processes for oversight of those activities, such as document and record control, internal audits, management review. It requires processed to ‘fix’ problems (i.e. ‘corrective actions’) for ‘nonconforming product’, complaints, and adverse events. It also defines the records and reports that must be maintained and can be inspected by the FDA.
Part 821: Medical Device Tracking Requirements
Requires certain medical device manufacturers (mostly implanted devices) to keep records of who is using their device in case they need to recalls or fix issues. Only applies to IVDs where a failure could have ‘serious adverse health consequences’.
Part 822: Postmarket Surveillance

The FDA may require studies to monitor the performance of a IVD even after it is approved and marketed. These rules describe the plan needed and the responsibilities of the sponsors and investigators of these studies.
Part 830: Unique Device Identification

Rules for a system to identify each IVD through distribution and use. The idea was to help customers and the FDA identify devices and monitor the device markets for safety issues. Each IVD must have a code (i.e. a ‘unique device identifier’) that identifies the IVD and production lot that can be read or scanned. Multiple agencies can issue the first part of the code, which is also submitted to a Global Unique Device Identification Database (GUDID).
Part 860: Medical Device Classification Procedures

These rules describe how medical devices are categorized and classified by risk (e.g. Class I, II, or III) and how each category of medical device is identified. This process can include advisory committees and classification panels. Each category may then be given specific regulations for that type of medical device. See section on Parts 862-892 below for more info). The de novo process allows manufactures of potential Class II IVDs with no similar test on the market to go through a streamlined process for categorization and classification.
Part 861: Procedures for Performance Standards Development

The rules describe how the FDA may create performance standards for class II and III medical devices. In the case of an IVD, this might include standards for how to perform analytical validation. These standards can then be referenced and consider mandatory in the sub-regulations specific to each category (type) of medical device. In this way, the FDA can simply update a standard without updating multiple regulations.
Parts 862-892: [Rules specific to a category or sub-category of devices.]
In these parts, the FDA gets into rules specific to just a subset of devices. The rules are divided into Parts for high-level categories like ‘Part 866 Immunology and Microbiology Devices’, then subparts like ‘Subpart B Diagnostic Devices’.

Each subpart is then divided into categories of tests like ‘§ 866.1645 Fully automated short-term incubation cycle antimicrobial susceptibility system.’

Lastly, the regulation identifies the category, gives a classification (I, II, or III) and may list additional exemptions or rules (i.e. ‘special controls’) such as labeling, submission information, testing (i.e. ‘design verification and valdiation’), guidance documents

Part 895: Banned Devices
Lists medical devices the FDA explicitly prohibits.
Part 896: Performance Standard for Electrode Lead Wires and Patients Cables
A performance standard the FDA decided to give its own part.
Other Parts You Need to Know
Part 11: Electronic Records and Signatures

Rules for computer systems. Electronic records and electronic signatures need to be follow these rules to be considered trustworthy, reliable, and equivalent to paper records. Rules cover the creation, modification, maintenance, archiving, retrieval, and transmission of electronic records under any FDA regulations (including medical device regulations). It describes computer system testing (‘validation’) audit trail requirements, and record retention for electronic records.
Recent Comments