I am a regulatory affairs and quality assurance professional in the in vitro diagnostic space. I have a Masters in Biomedical Regulatory Affairs from the University of Washington, and am RAC-device certified. I host this website in my free time, and provide all content free-of-cost. Please note that the content on this website is for informational and educational purposes only, and does not constitute legal or professional advice. All content and opinions shared are solely my own, and not affiliated with any company or organization.
I am a regulatory affairs and quality assurance professional in the in vitro diagnostic space. I have a Masters in Biomedical Regulatory Affairs from the University of Washington, and am RAC-device certified. I host this website in my free time, and provide all content free-of-cost. Please note that the content on this website is for informational and educational purposes only, and does not constitute legal or professional advice. All content and opinions shared are solely my own, and not affiliated with any company or organization.
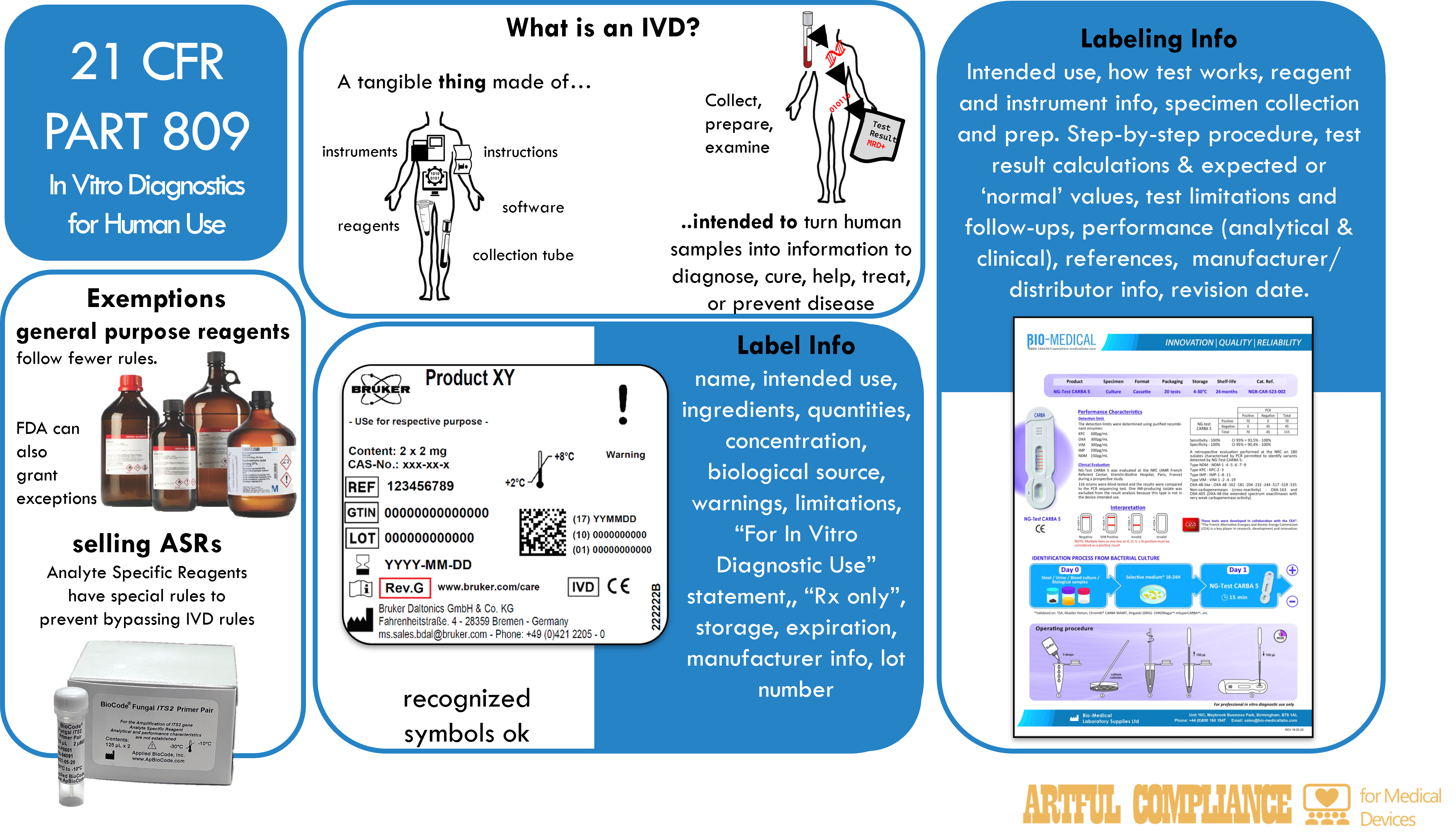
This regulation contains the labeling requirements for in vitro diagnostic products, which are devices or biological products used for testing specimens from the human body. Due to their special nature, the labeling requirements are different than the general medical device labeling requirement. The focus of the regulation is the information that must be stated on the label and the accompanying labeling, but there are also rules about who certain IVDs can be sold to that attempt to put limitations on the use of Laboratory Developed Tests.
Part A. Definition of an IVD
The regulation begins by defining what an IVD is,
reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
Part B. Requirements for label and labelling of an IVD (809.10)
Labels vs Labelling
If you mistake the two, it will cause confusion and chaos in understanding the regulations.
Labels are the written, printed, or graphic matter on the immediate container (occasionally an outer container if there is a space constraint) of a medical device. Labels include tags, stickers, or other attachments to the product or its container.
Labeling is a broader term– it includes all labels, but also the package insert, which is a document that provides instruction for use and other information necessary for the user to fully understand the product, its use, and limitations.
Labels On the Product
The label for an in vitro diagnostic product must state the label info (see list below), and other important information as applicable. The label must be legible and appear on the immediate container unless the container is too small, in which case it can go on outer containers or wrappers. The purpose of the lot number is to allow tracing to full manufacturing history, including all sub-parts of a kit or instrument.
Label information:
(1) product name, (2) intended use, (3) reagents name ingredients, (3) quantity of the product or tests, (3) concentration of reactive ingredients, (3) biological source, (3) biological activity,(4) warnings, (4) the statement “For In Vitro Diagnostic Use”, (4) any other limitations, (4) for prescription products: “Rx only” or “Caution: Federal law restricts this device to sale by or on the order of a ___”, (e.g. “physician”), (5) storage instructions, (5) reconstitution instructions, (6) expiration date (or other indicators), (7) quantities individual reagents, (8) manufacturer info, (9) lot number, and other information as applicable.
Labeling Accompanying the Product
The labeling that comes with the product, such as a package insert, must state the information below, in order.
Labeling information:
(1) name, (2) intended use, (2) type of procedure, e.g., qualitative or quantitative, (3) Summary and explanation of the test, (4) chemical, physical, physiological, or biological principles of the procedure, (5) Reagent information, (6) Instrument information, (7) specimen collection and preparation information, (8) Procedure ( step-by-step, and any points that may be useful in improving precision and accuracy), (9) description of result and how to calculate them, (10) Limitation of the procedure, (10) results that indicate further testing is needed, (11) expected values (e.g. range), (11) how the range(s) was established (e.g. ‘normal’ population), (12) specific performance characteristics, (13) Bibliography and references, (14) manufacturer/packer/distributor info, (15) revision date.
This information is often lengthy, and as a result rather than printing each time, manufacturers provide a document that shares where the information is available on a website.
Exceptions or alternatives
The FDA can grant exceptions or alternative to any labeling requirement if compliance with the requirement could adversely affect the safety, effectiveness, or availability of the products. The request for an exception or alternative must be submitted in writing and include rationale.
General purpose laboratory reagents and equipment
Labels of general purpose laboratory reagents and equipment that are not intended for a specific diagnostic application are exempted if their labeling meets a small set of requirements: description, composition, purity, quality, warnings, storage instructions, quantity, and lot number of the product.
Analyte specific reagents (ASR)
The labels of analyte specific reagents (e.g. monoclonal antibodies, DNA probes, viral antigens, and ligands) is exempted if their labeling meets a small set of requirements: for example name, quantity, purity, quality, warnings, storage instructions, expiration date, manufacturer, lot number, and the statement “Analyte Specific Reagent. Analytical and performance characteristics are not established” or “Analyte Specific Reagent. Except as a component of the approved/cleared test (Name of approved/cleared test), analytical and performance characteristics are not established”.
ASR labeling must not make any statement regarding analytical or clinical performance. The laboratory that develops an in-house test using the analyte specific reagent must inform the ordering person of the test result by appending the statement “This test was developed and its performance characteristics determined by (Laboratory Name). It has not been cleared or approved by the U.S. Food and Drug Administration.”
Use of symbols
The labeling of in vitro diagnostic products may use symbols in place of text if
the symbol is accompanied by explanatory text adjacent to the symbol, or
the symbol is contained in a standardrecognized by the FDA, or
the symbol is established by a standards development organization and is determined by the manufacturer to be likely to be read and understood by the ordinary individual.
The symbol must be used according to the specifications of the standard and must be explained in a symbols glossary that is included in the labeling.
Part C—Requirements for Manufacturers and Producers
IVDs are Medical Devices
This part is about selling IVDs. FDA reminds us that IVDs are medical devices, and sometimes biological products. As such, manufacturers of IVD products must comply with good manufacturing practices and, if applicable, biological product standards.
Selling Analyte Specific Reagents
There are also specific rules about selling analyte specific reagents (ASR’s). The general idea is that the FDA wanted to prevent ASR manufacturers from circumventing the 510K or PMA process by selling ASRs as RUO reagents to laboratories ad telling the labs they could use them as Laboratory Developed Tests. As a result, ASRs must be labeled and advertised according to specific requirements. Laboratories that use ASR’s to develop in-house tests must inform the ordering person of the test result and the test status.
Over-the-counter test sample collection systems for drug abuse testing
There are special rules for labeling of over-the-counter (OTC) test sample collection systems for drugs of abuse testing. These are restricted devices that must ensure sample testing is performed in a laboratory using approved or recognized screening tests. The labeling of these devices must provide adequate instructions and information for the lay users.
Have you ever wondered why each State doesn’t have their own laws about medical devices?
21 CFR Part 808, for the most part, preempts (i.e. prevents) states and localities from making their own rules about medical devices, except under certain conditions (exemptions). It provides procedures for the submission, review, and approval of applications for exemption from federal preemption by the FDA.
If preemption is an unfamiliar concept, consider this metaphor.
Imagine a country is represented by a family of four; two parents, a son, and a daughter.
The parents represent the Federal government.
The son and the daughter represent States governments who have to follow the rules.
The parent’s rules are like federal laws and regulations; they apply to everyone in the family and override any other rules that conflict with them. For example, the parents may have a rule that says no one can eat ice cream before dinner. This rule is the same for everyone and cannot be changed by anyone else.
The son’s and the daughter’s rules are like the State laws. They apply only to themselves or their own rooms. For example, the son may have a rule that says he can play video games for an hour every day. This rule is different from the father’s and the mother’s rules, but it does not contradict them, so it is valid.
If the daughter wants a rule that says she can eat ice cream before dinner, this rule contradicts the father’s rule, so it is not valid. This is called preemption. The parent’s rule preempts the daughter’s rule and makes it invalid.
Exemption from Preemption
But what if the daughter has a good reason to have her own rule? For example, what if she has a medical condition that requires her to eat ice cream before dinner? In that case, she may ask the parents for an exemption from their rule.
An exemption is an exception that allows a State to have a different rule that would otherwise be preempted. The parent (Federal government) may grant the exemption if they agrees that the daughter has a valid reason and that her rule is more beneficial for her health or safety. For example, the parent may say that the daughter can eat ice cream before dinner, but only if she has a doctor’s prescription and only a small amount. This is called an exemption from preemption. The parent’s rule still applies to everyone else, but the daughter has a special permission to have her own rule.
To apply for an exemption, a state or locality must send an application to the FDA with various information about the device, the requirement, and the reason for the exemption. The FDA will review the application and publish its decision in the Federal Register, and may revoke the exemption if necessary.
Examples of Exemption From Preemption for Medical Devices
One example of exemption from preemption is the California Radiation Control Law, which regulates the use of radiation-emitting devices, such as X-ray machines, in the state. The FDA granted an exemption from preemption for this law in 1980, because it found that the state law was more stringent than the federal performance standards for radiation-emitting devices, and that it addressed a specific local problem of excessive radiation exposure.
Another example of exemption from preemption is the New York State Department of Health’s requirement for premarket approval of HIV home test kits, which are devices that allow individuals to test themselves for HIV infection at home. The FDA granted an exemption from preemption for this requirement in 1996, because it found that the state law was more stringent than the federal premarket approval process for HIV home test kits, and that it served a compelling local need to ensure the accuracy and reliability of such devices.
In my opinion, registration and listing is the simplest and most intuitive regulation for medical devices. It’s a quick and fairly painless online process if you read and follow FDAs detailed instructions. Device Registration and Listing | FDA
The FDA is simply trying to keep track of the medical devices that are produced or imported into the U.S. Knowing who is making what is an obvious precursor step to monitor manufacturers, identify potential problems, and contact them as needed.
Registration & Listing
If you can remember the following three rules, you will be in good shape.
Rule 1: ‘Establishments’ must register and list their devicesunless they get a waiver.
Don’t get your hopes up– you probably don’t qualify for a waiver.
Establishment means a place of business under one management at one general physical location at which a device is manufactured, assembled, or otherwise processed
But please note that ‘establishments’ include a far wider scope than you might guess, including locations that design and develop products, repackage or relabel, or sterilize: Full list of activities here: Domestic Establishments | FDA
Rule 2: Establishments must review and update registration and listings annually
You will be required to pay a fee up front for this privilege.
Rule 3: Establishments must update their registration and listing information within 30 days of certain changes
This includes changes to…
name, address, or ownership of the establishment.
devices with activities at the establishment
activities that are performed(e.g. design, manufacturing, repackaging, etc.).
status of the devices (commercially distributed, discontinued, or resumed)
premarket submission number (510 (k), De Novo, PMA, PDP, HDE) for the devices
importers and persons who import or offer for import the devices
NOTE: This blog covers only 21 CFR Part 807 subparts A to D. Part E covers premarket notifications (a type of submission) and will be described in a later blog.
‘Recall’ of a medical device is when a manufacture takes action to address a problem with a device that violates FDA law OR has a defect that could be a risk to patient healt.. The action may be to repair, modify, adjust, relabel, inspect, or destroy it. They come in two flavors depending on wherethe action happens 1) Correction and 2) Removal.
‘Correction’ is when the action to address the problem occurs without physically removing it from where the device is used or sold. It’s not always feasible or necessary to take back a device to fix an issue. A device might simply need to be checked or adjusted by a technician, or new software installed. Sometimes a correction is just to provide new instructions for use.
‘Removal’ is when the action to address the problem is to physically remove it from where it is used or sold.
History of the Recall Regulation
Before the Safe Medical Devices Act of 1990 (SMDA), reporting recalls of medical devices was voluntary. It was like letting students decide which exams to turn in—the teachers are unlikely to get an accurate picture of student performance or know how to help failing students. Congress believed there was under-reporting of recalls to the FDA. In turn, this left the FDA unable to do its job of evaluating device-related problems and how those problems were being corrected.
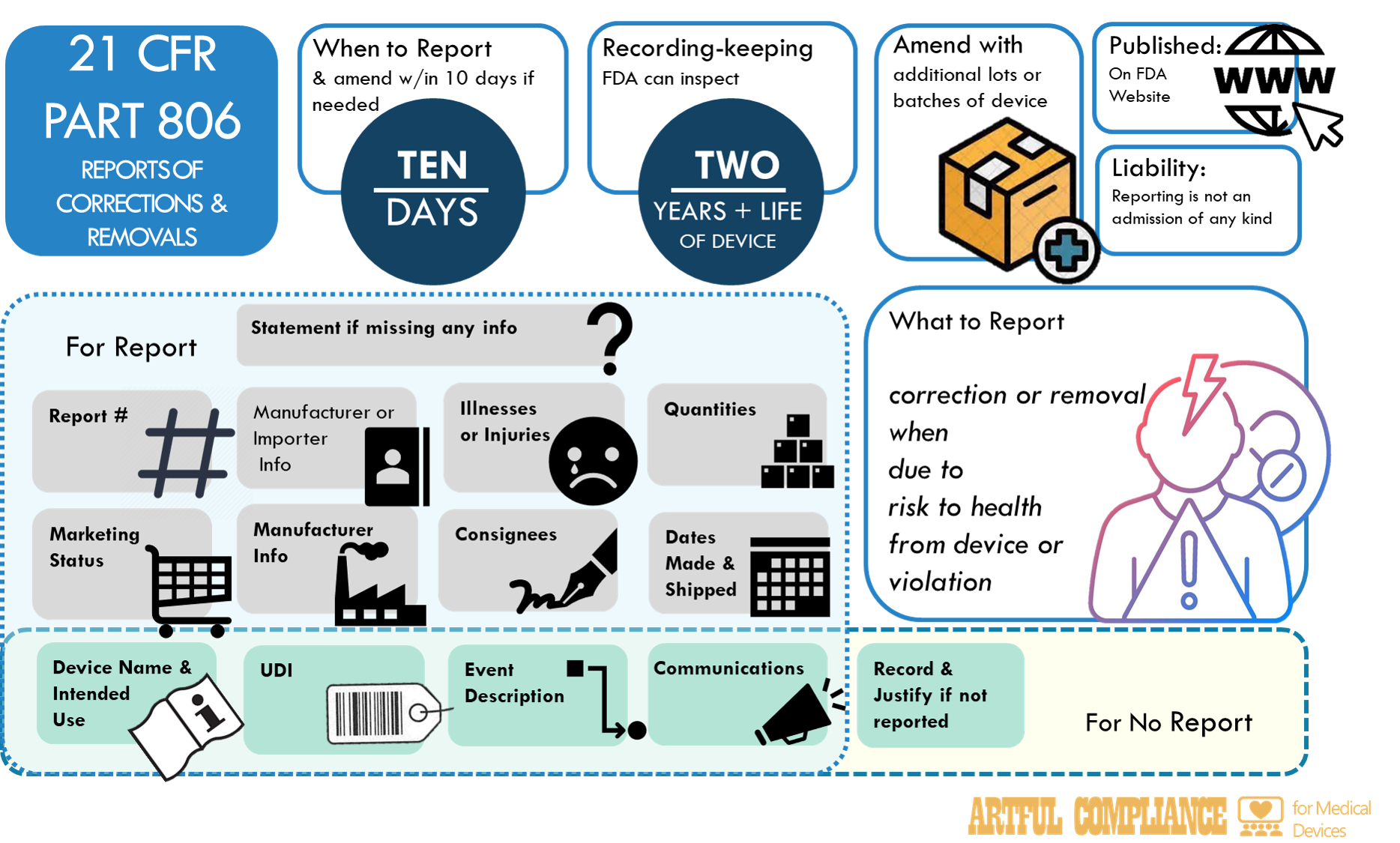
After the SMDA, the FDA created 21 CFR Part 806, Reports of Corrections and Removals. It mandated manufacturers (including Ex-US) to promptly report certain types of device corrections and removals to the FDA. Manufacturers also had to maintain records of all corrections and removals regardless of whether they met the criteria for reporting to the FDA. 21 CFR Part 806 is a scant 4 pages long, and not a challenging read, but here’s the top things you should know.
Not Everything Needs to Be Reported
There are some recall actions you don’t need to report (21 CFR 806.1(b)):
Improvements not related to health risks or violations
Actions that improve performance or quality of a device but do not reduce a risk to health posed by the device or remedy a violation of device laws. (see below for definition of ‘risk to health’)
Market withdrawal
Actions that involve a minor violation of the act that would not be subject to legal action by FDA or that involves no violation of device laws.
Routine servicing
Any regularly scheduled maintenance of a device (e.g. planned part replacement, calibration, battery replacement), and responses to normal wear and tear. Unexpected repairs or replacements or unexpected frequency of this activity are not routine servicing.
Stock recovery or rotation
If a device has not been marketed or has not left the direct control of the manufacturer, i.e., is located on the premises owned, or under the control of, the manufacturer, and no portion of the lot, model, code, or other relevant unit involved in the recall been released for sale or use.
Actions already reported as part of Adverse Event
If you’ve already reported the recall in following the adverse event reporting (see 21 CFR Part 803) process, you don’t need to do it again.
Of particular interest to LDT laboratories will be the stock recovery exemption. It means that even if a lot of reagents has been released to the laboratory, so long as the laboratory can show the lot has not been used on patient samples, it can be argued that it can be removed from the lab without considering it a recall.
Recalls to Reduce A Risk To Health Must be Reported
A lot of the decision to report or not hinges on the words ‘risk to health’, so let’s explore that a little.
Risk to health means (1) A reasonable probability that use of, or exposure to, the product will cause serious adverse health consequences or death; or(2) That use of, or exposure to, the product may cause temporary or medically reversible adverse health consequences, or an outcome where the probability of serious adverse health consequences is remote
Medical device manufactures of typical IVDs know risk management pretty well— it is baked into ISO 13485, and ISO 14971. But laboratory professionals responsible for LDTs may not be as familiar with definitions of risk. In simple terms, “risk” considers probability (likelihood) and severity (consequences). The FDA defines three ‘classes’ (I, II, III) due to use of a device that constitute a reportable risk to health:
Situation
Probability
Severity
Class I
Reasonable
Serious adverse heath consequences or death
Class II
Possible (“may”)
Temporary or medically reversible
Class II
Remote
serious adverse health consequences
Class III (voluntary to report)
Not likely
adverse health consequences
This means that to determine if there is a ‘risk to health’ your team must first estimate (using available data) both the probability and severity of problem with the IVD, and depending on that evaluation determine if the problem meets the criteria in this table. If you aren’t sure, you should report, and keep in mind that the FDA will makes its own evaluation (aka a ‘health hazard assessment” to classify your recall.
Report Promptly
The FDA didn’t leave what this means up to the manufacturer. Promptly means within ten working days of starting the recall.
How to Report
The FDA doesn’t have a form for reporting recalls, but requires manufactures to include all of the following (21 CFR 806.10(c)):
Report number: registration, date, sequence, and type
Manufacturer or importer: representative info
Device name and use
Device marketing status: number
Device ID: UPC, model, catalog, code, lot, or serial number
Manufacturer info: if different from reporter
Event and action: description
Illness or injuries: report numbers if any
Number of devices and batches: for correction or removal
Manufacture or distribution date: device life
Consignees info: distribution details
Communications copy: recipients not in element
Reason and date: for missing information
Moreover, if you end up extending recall to other products, you have to ‘promptly follow up with another report amending the original report. You have two ways to get this info to the FDA
As with adverse event reporting, reporting a recall does not mean the manufacturer is admitting their device caused illness or injury. The report is legally protected from being used in that way.
Record all Recalls, Even the Non-Reported Ones
Even if the recall actions don’t need to be reported, you must maintain records of those actions. The list of things to record is less than for reporting and includes:
Brand name, classification, name and product code if known, and the intended use of the device.
Identification. Unique device identifier (UDI) of the device, or other unique identifier code and model/catalog #, and lot or serial # if known.
Description of the event(s) giving rise to the information reported and the corrective or removal action that has been, and is expected to be taken.
Justification for not reporting the correction or removal action to FDA, including conclusions and any followups, and be reviewed and evaluated by a designated person.
Copy of all communications regarding the correction or removal.
The FDA is allowed to (and will likely) inspect these records in the future.
A recall is a voluntary action that can be taken by a manufacturer, or at the FDA’s request. It’s almost always a better choice than seizure or other court actions. Make no mistake– the FDA can and will use force if a a manufacturer does not voluntary recall devices to their satisfaction. The process by which the FDA evaluates reported recalls, classifies recalls, asks for or mandates a recall, or decides whether the manufacturer’s recall plan is good enough is described in 21 CFR 7. In also discuss the more police-like actions the FDA can take. This will be the topic of another blog.
This regulation describes rules for reporting adverse events to the FDA and states how these reports may be shared with the public. Adverse events include, but are not limited to, patient harm due to incorrect results as a result of IVD malfunctions, or faulty user instructions or labelling.
Future Applicability to LDTs in Stage 1
If the FDA gets its way, Laboratory Developed Tests (LDTs) will soon become FDA-regulated in vitro diagnostic (IVD) medical devices. In the recently proposed rule, Stage 1 of the transition of LDTs to IVDs would require LDTs to follow medical device reporting rules in 21 CFR Part 803. Labs with LDTs will need to brush up on 21 CFR Part 803, Medical Device Reporting (MDR).
CLIA labs are already user facilities & must report adverse events for medical devices… …but LDTs are not currently medical devices
The MDR regulation requires manufacturers, importers, and user facilities to report certain adverse events and product problems involving medical devices to the FDA. If you are a clinical diagnostic laboratory under CLIA, you may be surprised to learn that you are already obligated to report adverse events for medical devices. This is because user facilities include outpatient diagnostic facilities that conduct medical diagnostic tests on patients using in vitro testing (i.e. CLIA labs). See 21 CFR 803.3(r) for more info.
But the key words is adverse events for medical devices. Because a LDT is not a ‘medical device’ or at least the regulation hasn’t ever been enforced), adverse events for LDTs do not need to be reported to the FDA. Yet.
Once LDTs become IVDs, CLIA labs will be both manufacturers and user facilities of IVDs
It’s a dummy whammy. Once LDTs are no more, CLIA labs will now be both making and using an IVD.
They will need to start reporting adverse events to the IVD manufacturer as a user facility. AND
They will need to start reporting adverse events to the FDA as an IVD manufacturer.
Still confused? Look at it this way. In a traditional Manufacturer-CLIA lab relationship, this how adverse events would play out:
Step
Traditional Model
LDTs are Now IVDs Model
Design & Manufacturer
The manufacturer designs and makes an IVD and sells it to a CLIA lab.
The CLIA lab designs and makes an IVD and makes it available to its own CLIA lab.
Use Leading to Adverse Event
The CLIA lab uses the IVD and learns it has a design issue or has malfunctioned. For instance, the IVD has a serious analytical performance issue (such as those caused by poor reagent lot or software error), or clinical performance issue (e.g. the test results in incorrect clinical decisions). Due to these issues, a test result is acted upon by a physician, leading to an adverse event.
User Facility Reporting[FORM 3500A w/in 10 days for death or serious injury, and FORM 3419 annually]
The CLIA laboratory (as a user facility) reports an adverse event to the manufacturer AND in the case of death, reports the adverse event to the FDA.
The CLIA laboratory assesses both as a user facility & a manufacturer whether there has been an adverse event (death, serious injury) or malfunction, and reports the adverse event to the FDA.
Manufacturer Reporting[electronic MDR w/in 30 days for death, serious injury, and malfunctions, 5 days for events needing remedial actions]
The manufacturer independently assesses whether there is a reportable adverse event, and reports it to the FDA.
You might think the LDT model is simpler; after all, it cuts out the middle-man in deciding if something is an adverse event. But this does raise some questions and issues.
When the CLIA Lab is the User Facility & Manufacturer, what is the timeline for reporting? Can each function report separately?
According to 21 CFR 803.3(b), you ‘become aware’ of an event when an employee of the reporting entity has information that reasonably suggests that a reportable adverse event occurred.
This means that a CLIA lab may be a ‘reporting entity’ that has employees both responsible for reporting adverse events as a user facility, and an employees responsible for reporting adverse events as a manufacturer. The manufacturer therefore may ‘become aware’ as soon as the user facility does.
Unless you allow the definition of ‘reporting entity’ to split the internal functions within a single organization into ‘manufacturer’ and ‘user’ this significantly compresses the time for external reporting to the FDA.
30 days (Manufacturer) – up to 10 days (User Facility) = 20-30 days for Manufacturer
versus
30 days (Manufacturer) + 10 days (User Facility) = 30 days for Manufacturer
Essentially, any time it takes for the user facility to report the adverse event to the manufacturer is now subtracted (up to ten days) from the manufacturer’s time, giving 20 days to evaluate and report to the FDA.
And what about when the adverse event is death? The User facility is obligated to report that to the manufacturer AND the FDA within 10 days. Is the assumption then that the manufacturer follows up with more information about the device within 30 days AFTER that?
But does that mean the user and the manufacturer file separate reports, even though they are the same entity? Maybe, the FDA would seem to allow separate reports, but hypothetically, you could also combine into a single report. The regulations and guidance do not address a scenario where a single entity has these dual roles.
Are Adverse Events Reportable if they are ‘caused or contributed’ by User Errors?
According to 21 CFR 803.3(c) a device has ‘caused or contributed’ to an event if a death or serious injury was or may have been attributed to the device, or if the device was or may have been a factor in a death or serious injury, including events occurring as a result of: (1) Failure, (2) Malfunction, (3) Improper or inadequate design, (4) Manufacture, (5) Labeling, or (6) User error.
Laboratory errors are mistakes or inaccuracies that occur during the process of testing, analyzing, or reporting medical samples in a laboratory setting. These errors include examples such as errors in sample receipt or handling, adding the wrong reagent, reagent storage errors, selecting an incorrect instrument program, sample swaps, or QC errors. Laboratory errors can have serious consequences for patients, such as misdiagnosis, delayed treatment, or inappropriate interventions. Are these ‘user errors”?
Per the FDA “A use error refers to a situation in which the outcome of device use was different than intended, but not due to malfunction of the device. The error may have been due to a poorly designed device, or it may have been used in a situation that promoted incorrect usage. Other users may make the same use error with similar or worse consequences.”
Thus, one interpretation of the requirement to address adverse events that are a result of ‘user error’ would be limit this to laboratory errors due to using the IVD in a way that clearly different than the manufacturer intended, but inherent to the design of the IVD. Examples might be where the IVD reagents or software have inherent flaws that are likely to lead the CLIA lab staff to improper use. This is likely to be a small subset of all laboratory errors, and not any or all mistakes made by lab staff when performing a testing procedure.
However, I have seen labs take the view that all lab errors are ‘user errors’ of an IVD. If this is your interpretation, I strongly recommend you involve a Legal representative in that decision. There is currently a lack of incentives and protections for reporting laboratory errors, such as confidentiality, anonymity, immunity, or feedback, and the issue of legal liability, professional sanctions, or reputational damage will be points for discussion.
Complaint Files and Medical Device Reporting
It is impossible to discuss Medical Device Reporting, without also discussing complaints. Complaint files are linked to MDR event files because a complaint must be evaluated to determine if it is a reportable adverse event. A complaint is:
“any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution”.
Manufacturers are required to maintain complaint files and establish and maintain procedures for receiving, reviewing, and evaluating complaints. Complaint files that are found to be reportable MDR events should be maintained in a separate portion of the complaint file or otherwise clearly identified.
It should be noted that CLIA laboratories are also required “have a system in place to ensure that it documents all complaints and problems reported to the laboratory. The laboratory must conduct investigations of complaints, when appropriate.” See §493.1233 Standard: Complaint investigations
If the CLIA lab is also the IVD manufacturer, are they required to evaluate ANY issues identified in laboratory records, or in discussion with laboratory staff?
This is a problematic question for both adverse events, and complaints in general. As a single entity, the manufacturer may now have access to quality records of the laboratory, including all complaints from laboratory customers. The development or manufacturing team may also be in direct contact with laboratory staff using the test on a routine basis. As such, the manufacturer could ‘become aware’ of a wide variety of customer and internal lab user issues that could constitute a complaint.
After ‘becoming aware’ they may now think they need to assess each issue for whether it could be related to the labeling or user error as a complaint, and then as a potential malfunction or adverse events.
This is problematic in three ways.
A) potentially every lab event or corrected report could be a candidate for evaluation by the ‘manufacturer’.
B) the design and manufacturing team generally does not have the same experience and understanding of laboratory workflows to evaluate lab errors.
C) Even if the design and manufacturing team could evaluate all issues, the authority and responsibility to do so is designated to the Clinical Laboratory Director (CLD), under 42 CFR Part 493 (CLIA), not to the manufacturer under 21 CFR Part 820 or 803.
A solution to this conundrum is proposed below.
Separation of User and Manufacture in the Adverse Event Reporting Process
My recommendation from experience is to not make the ‘manufacturing’ part of the organization directly responsible for evaluation of all laboratory issues, but instead to clearly separate the responsibilities within the organization as ‘manufacturer’ versus ‘user facility’ (i.e. the CLIA laboratory).
This would in practice mean to appoint a function (a person or group) within the CLIA laboratory as a separate complaint and adverse event evaluation team for the CLIA laboratory. Their purpose would be to record all complaints (and possible adverse events) to a smaller list of complaints and adverse events that are about the device as opposed to the laboratory service. Complaints would include feedback from their own laboratory users about device issues. This filtered set of would then be sent forward to a separate device ‘manufacturer’ team for evaluation.
The manufacturing team would be a separately appointed function that would then evaluate this pared down list of ‘device’ complaints for adverse events and to complete the more formal steps required by 21 CFR Part 820 (Quality System Requirement) for complaint investigation and reporting.
If the same team must be is used for both functions, there should be a clear division of he these steps of evaluation, to not confuse evaluation of ‘use’ with evaluation of ‘design’ or ‘manufacturing’.
This approach is further described below.
Step 1: The User Facility Determines if there is an Adverse Event
If the complaint / adverse event is being reported to the CLIA laboratory that tested a sample, it is being reported to the user facility. The user facility is then responsible for a first pass of the evaluation. The user facility responsible person (for instance the Clinical Laboratory Director, or their designee) then should determine if an adverse event has occurred due to a medical device. They may do this by asking if there is a reasonable possibility that;
a device failure
a device design defect , or
the device labeling
was a direct or indirect factor in a death or significant injury?
Its important to note that not just anyone can make this decision. It needs to be made by “a person who is qualified to make a medical judgment (e.g., a physician, nurse, risk manager, or biomedical engineer) to reach a reasonable conclusion.” It should also be noted, that the FDA rejected a suggestion that MDR reports be required only if the device is a significant factor in causing an adverse event. This is an important distinction in the case of an IVD, where the patient results is almost certainly and indirect rather than direct cause of an event.
If the answer is ‘No’, under CLIA they must still investigate and evaluate whether the lab needs to take corrective action. If the complaint or adverse event was due to laboratory error, they must investigate and correct the error per 42 CFR §493.1233 and 493.1239. But no adverse event reporting is required. The Information which leads the qualified person to determine that a device-related event is or is not reportable must be contained in the MDR event files, even if the event is not reported.
Step 2: The User Facility Reports the Adverse event to the Manufacturer, and FDA (in case of death)
If the event is a reportable adverse event in Step 1, under 21 CFR Part 803, they must report, within 10 days to the internal team within their company responsible for the ‘manufacturer’ role. A good choice for this is the Complaint Handling Unit (CHU) that the manufacturer will need to designate anyway per 21 CFR Part 802.198. It is strongly recommended that team is a different than the team in step #1, to prevent a misunderstanding of the role of each team.
Step 3: The Manufacturer Determines if there is an Adverse Event
Next, the group responsible for manufacturing will need to evaluate the adverse event. Again, they will ask the question if there is a reasonable possibility that;
a device failure,
a device design defect , or
the device labeling
was a direct or indirect factor in a death or significant injury?
Hypothetically, this might be a different answer than that decided by the CLIA laboratory, for instance because the CLIA laboratory may be more conservative about whether a laboratory error may be a ‘user error’ due to an inherent design flaw.
If the answer is ‘No’, as a manufacturer, this may still be considered a complaint, and require investigation and evaluation of whether the manufacturer needs to take corrective action. But no adverse event reporting is required.
Again, a qualified person must make the determinations and provide the information evaluated in the MDR event files, even if the event is not reported.
Step 4: The Manufacturer Reports the Adverse event to the FDA
If the answer is ‘Yes’, under 21 CFR Part 803, the manufacturer must report, within 30 days to the FDA. The steps for Electronic Medical Device Reporting (eMDR) and detailed FDA Guidance are on the FDA’s website.
Reporting Malfunctions
The user facility (the CLIA lab) may also decide to report to the group responsible for manufacturing that there was a device malfunction, even if it did not result in death or serious injury. Under 21 CFR Part 830, a user facility is not obligated to do so, but in the case a laboratory using the device is the same ‘reporting entity’ as the manufacturer they may be obligated as a ‘reporting entity’ to do so.
If the lab does report a device malfunction meeting this criteria, the manufacturer would then have 30 days to evaluate for themselves if it met the criteria for a device malfunction, and then report that malfunction to the FDA.
With the latest FDA proposed rule-making for IVDs, laboratory professionals supporting Laboratory Developed Tests (LDTs) may be asking, what does this mean for my LDT test?
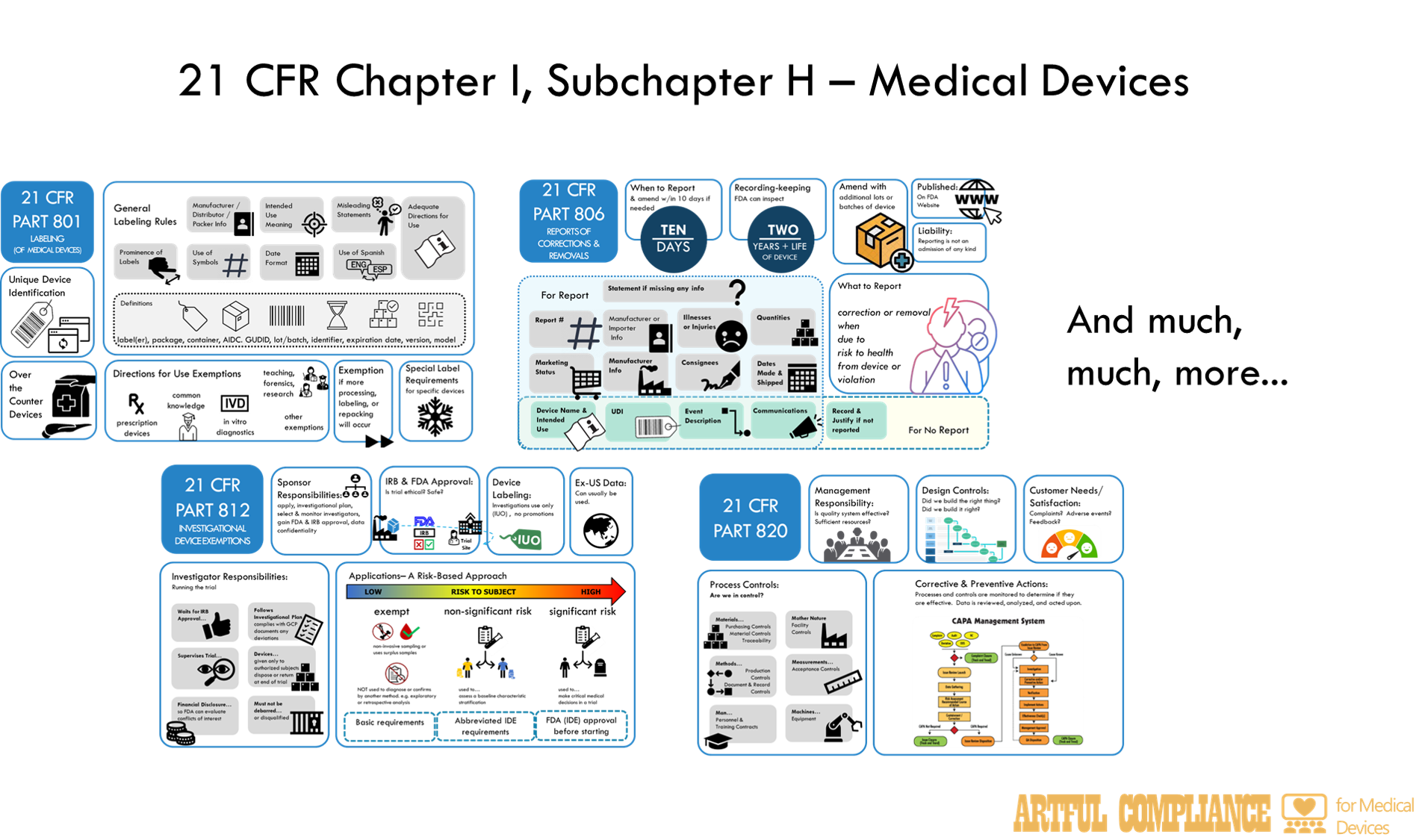
In short—the FDA wants to regulate LDTs under the same rules as in vitro diagnostic (IVD) medical devices. Those rules are spelled out in 21 CFR Chapter I, Subchapter H – Medical Devices. If you are not a Regulatory Affairs or Quality Assurance professional in the traditional medical device industry (and maybe even if you are), even looking at the table of contents may be a little daunting.
The aim of this post is to make it less so. I do this by describing each part in simple terms, and giving the relevance to a LDT, should LDTs start being regulated as IVDs. Once you have the high-level overview, you can dive down into each part. I plan to release more detailed information of each part in future blogs, so stay tuned. Note, I will omit information not applicable to IVDs to cut this information down.
Part 800: General
Administrative rules for how FDA will prevent selling or using devices they deem unsafe, and how to appeal those decisions.
Part 801: Labeling
Rules for ‘Labeling’ which includes far more than just physical labels on IVD reagents, but also instructions for the user of reagents, instruments, or software, plus any information provided to customers about what the IVD could or should be used for. This also includes promotional material or information provided in the patient result. Note that Part 809 provides more specific rules for labeling of IVDs.
Part 803: Medical Device Reporting
Rules for reporting adverse events to the FDA and states how these reports may be shared with the public. Adverse events include, but are not limited to, patient harm due to incorrect results as a result of IVD malfunctions, or faulty user instructions or labelling.
Part 806: Medical Devices; Reports of Corrections and Removals
Rule for reporting of corrections and removals of IVDs to the FDA. This is when a lab releases IVD reagents, instruments, or software to the lab to use in testing, and then later realizes there is something ‘wrong’ with them and needs to do something to ‘fix’ the problem or take the faulty parts of the IVD away from lab users.
Part 807 (A-D): Establishment Registrations & Device Listing
Rules for how and when to register the facilities (i.e. ‘establishment’) that designs and makes the IVD, and providing a list of IVDs (i.e. ‘listing’) it makes at each facility, to the FDA. This is public information and must be updated annually.
Part 807 (E): Establishment Registrations & Device Listing
Rules for premarket notification submissions (aka ‘510k’) by which the FDA can agree (i.e. ‘clear’) that a medium risk IVD is like (i.e. ‘substantially equivalent’) to an IVD already being sold. After looking at analytical and (sometimes clinical) validation data, they can agree it is safe and effective enough to be sold.
Part 808: Exemptions From Federal Preemption of State and Local Medical Device Requirements
Federal preemption means that FDA rules can override state rules if they conflict with each other. This rule gives States a path to have different rules if they can show their rules are stricter than the FDA’s.
Part 809: In Vitro Diagnostic Products for Human Use
Specific labeling rules for IVDs including call outs for; how to label general and analyte-specific reagents, labeling instruments, what needs to go into IVD lab procedures and over-the-counter instructions, and how to describe IVD performance characteristics (e.g. accuracy, precision).
Part 810: Medical Device Recall Authority
How the FDA has the authority to issue a recall or cease distribution order. This means they can make IVD manufacturers ask users to send purchased IVD materials back at the manufacture’s expense, or stop manufactures from providing IVD products.
Part 812: Investigational Device Exemptions
If the FDA has not cleared or approved an IVD, it can still be used in a clinical trial, but is considered an ‘investigational device’ and requires an ‘exemption’ to use it. The Sponsor and manufacturer also need to follow certain rules depending on risk involved in using the IVD in the trial. For the highest risk category, a formal application to the FDA is needed.
Part 814: Premarket Approval of Medical Devices
Rules for premarket approval submissions (aka ‘PMA’) by which the FDA can agree (i.e. ‘approve’) that a high risk IVD is safe and effective enough to be sold. Both analytical and clinical validation data is generally needed, and there may be commitments to monitor the IVD or perform additional studies even after approval (i.e. ‘postapproval requirements’).
Rules for humanitarian IVDs for rare use cases (e.g. rare diseases).
This part explains how to get approval from the FDA for a new medical device that is not substantially equivalent to an existing one. It also describes the types of clinical studies that are required to demonstrate the safety and effectiveness of the device1.
Part 820: Quality System Regulations
please attribute to artfulcompliance.com
Rules for the quality system of IVD manufacturers, which includes procedures for design, production, packaging, labeling, storage, installation, and servicing of their devices. It requires processes for oversight of those activities, such as document and record control, internal audits, management review. It requires processed to ‘fix’ problems (i.e. ‘corrective actions’) for ‘nonconforming product’, complaints, and adverse events. It also defines the records and reports that must be maintained and can be inspected by the FDA.
Part 821: Medical Device Tracking Requirements
Requires certain medical device manufacturers (mostly implanted devices) to keep records of who is using their device in case they need to recalls or fix issues. Only applies to IVDs where a failure could have ‘serious adverse health consequences’.
Part 822: Postmarket Surveillance
The FDA may require studies to monitor the performance of a IVD even after it is approved and marketed. These rules describe the plan needed and the responsibilities of the sponsors and investigators of these studies.
Part 830: Unique Device Identification
Rules for a system to identify each IVD through distribution and use. The idea was to help customers and the FDA identify devices and monitor the device markets for safety issues. Each IVD must have a code (i.e. a ‘unique device identifier’) that identifies the IVD and production lot that can be read or scanned. Multiple agencies can issue the first part of the code, which is also submitted to a Global Unique Device Identification Database (GUDID).
Part 860: Medical Device Classification Procedures
These rules describe how medical devices are categorized and classified by risk (e.g. Class I, II, or III) and how each category of medical device is identified. This process can include advisory committees and classification panels. Each category may then be given specific regulations for that type of medical device. See section on Parts 862-892 below for more info). The de novo process allows manufactures of potential Class II IVDs with no similar test on the market to go through a streamlined process for categorization and classification.
Part 861: Procedures for Performance Standards Development
The rules describe how the FDA may create performance standards for class II and III medical devices. In the case of an IVD, this might include standards for how to perform analytical validation. These standards can then be referenced and consider mandatory in the sub-regulations specific to each category (type) of medical device. In this way, the FDA can simply update a standard without updating multiple regulations.
Parts 862-892: [Rules specific to a category or sub-category of devices.]
In these parts, the FDA gets into rules specific to just a subset of devices. The rules are divided into Parts for high-level categories like ‘Part 866 Immunology and Microbiology Devices’, then subparts like ‘Subpart B Diagnostic Devices’.
Each subpart is then divided into categories of tests like ‘§ 866.1645 Fully automated short-term incubation cycle antimicrobial susceptibility system.’
Lastly, the regulation identifies the category, gives a classification (I, II, or III) and may list additional exemptions or rules (i.e. ‘special controls’) such as labeling, submission information, testing (i.e. ‘design verification and valdiation’), guidance documents
Part 895: Banned Devices
Lists medical devices the FDA explicitly prohibits.
Part 896: Performance Standard for Electrode Lead Wires and Patients Cables
A performance standard the FDA decided to give its own part.
Other Parts You Need to Know
Part 11: Electronic Records and Signatures
Rules for computer systems. Electronic records and electronic signatures need to be follow these rules to be considered trustworthy, reliable, and equivalent to paper records. Rules cover the creation, modification, maintenance, archiving, retrieval, and transmission of electronic records under any FDA regulations (including medical device regulations). It describes computer system testing (‘validation’) audit trail requirements, and record retention for electronic records.
The FDA has published a proposed rule to amend 21 CFR Part 809 (in vitro diagnostic) regulations to state that laboratory developed tests (LDTs) are in vitro diagnostic products (IVDs) designed, manufactured and used within a single clinical laboratory, and need to follow the same rules as other medical devices.
What you should know:
The FDA believes LDTs are IVDs and it’s going to regulate them as such. This is a point that should come as no surprise as the FDA has made it ad nauseum for going on 20 years now. The FDA’s oft-repeated line is that in the almost 50 years since the 1976 Medical Device Amendments, it has simply been practicing ‘enforcement discretion’. In other word’s it has simply decided not to do anything about the thousands of labs not following the IVD regulations, but always maintaining they could if they felt it was needed.
The FDA has a long (unsuccessful) history of trying to start enforcing IVD rules for LDTs. It started 20+ years ago. Jeffery Gibbs provides a comprehensive review of this in LDTs: The Saga Continues. The whole thing reads like two neighbors arguing over a property line. Industry, and especially academic medical centers have fought the enforcement tooth and nail. The latest failure to get the VALID act through Congress appears to have been the FDA’s last straw.
The FDA expects a legal battle. Using the ‘rule-making process’, to call LDTs as IVDs effectively bypasses Congress and goes straight into the process to simply update the regulation (21 CFR Part 809). It’s the equivalent of saying the property line is ten feet over and simply starting the process to move the fence. Since the VALID act went to Congress in the first place, that would seem to imply the FDA knows Congressional action was needed to change the regulation. The challenge of FDA’s legal authority is bound to be a great show. Bring the popcorn.
Exemptions and Exceptions. The rule provides a smorgasbord of labs and tests that may be exempt (e.g. blood typing, forensics), might have less stringent rules (academic labs, low revenue labs, tests approved by New York or the VHA), or that might get additional transition time. It asks for feedback from the public on all of these possibilities, in what will almost certainly be a very long comment review process.
Benefits & Costs. Per the FDA, the rule is aimed at helping to ensure the safety and effectiveness of LDTs and improving innovation. Most of the 83 page document in the Federal Register are devoted to why the FDA believes LDTs are unsafe. Sure, improving patient safety is great. But labs are nervously eyeing the price tag. The FDA outlines benefits ($86B/year) and costs of the rule (5.8B to labs, plus 500M to FDA that will be passed off to labs as user fees). It’s anyone’s guess what magic hat the FDA pulled those estimates from. The Pew Trust researched the topic in 2021 and concluded it was ‘difficult to know precisely how many LDTs are on the market, or to accurately estimate the volume of tests that are run using LDTs [but] they are clearly common, and many labs rely on them in some capacity.”
Timeline. As you might expect, the FDA’s plan is to phase in the new approach. See graphic below. Year one starts with adverse event reporting and the ability to recall IVDs. Year two will require registration and listing. Year three will bring QMS requirements (design and manufacturing controls, CAPAs, complaints, etc under 21 CFR Part 820) which by that time might actually be updated to align with ISO 13485. Lastly, year 3.5 and 4 will mark deadlines for FDA application for Class III PMA and Class I/II de novos or 510Ks.
What Does the Future Hold? Considering the concerns that the FDA can’t even handle its current workload with IVDs, no doubt these cut-offs will be followed with a lengthy period of years in which the absolutely overloaded FDA will wade through a backfill funded by astronomical user fees that will still not cover the work that needs to be done as inexperienced labs take on their first FDA applications. If you are a lab with one or more LDTs, the future holds much uncertainty. If you are a regulatory affairs professional in the IVD industry, expect job security and a raise.


























Recent Comments