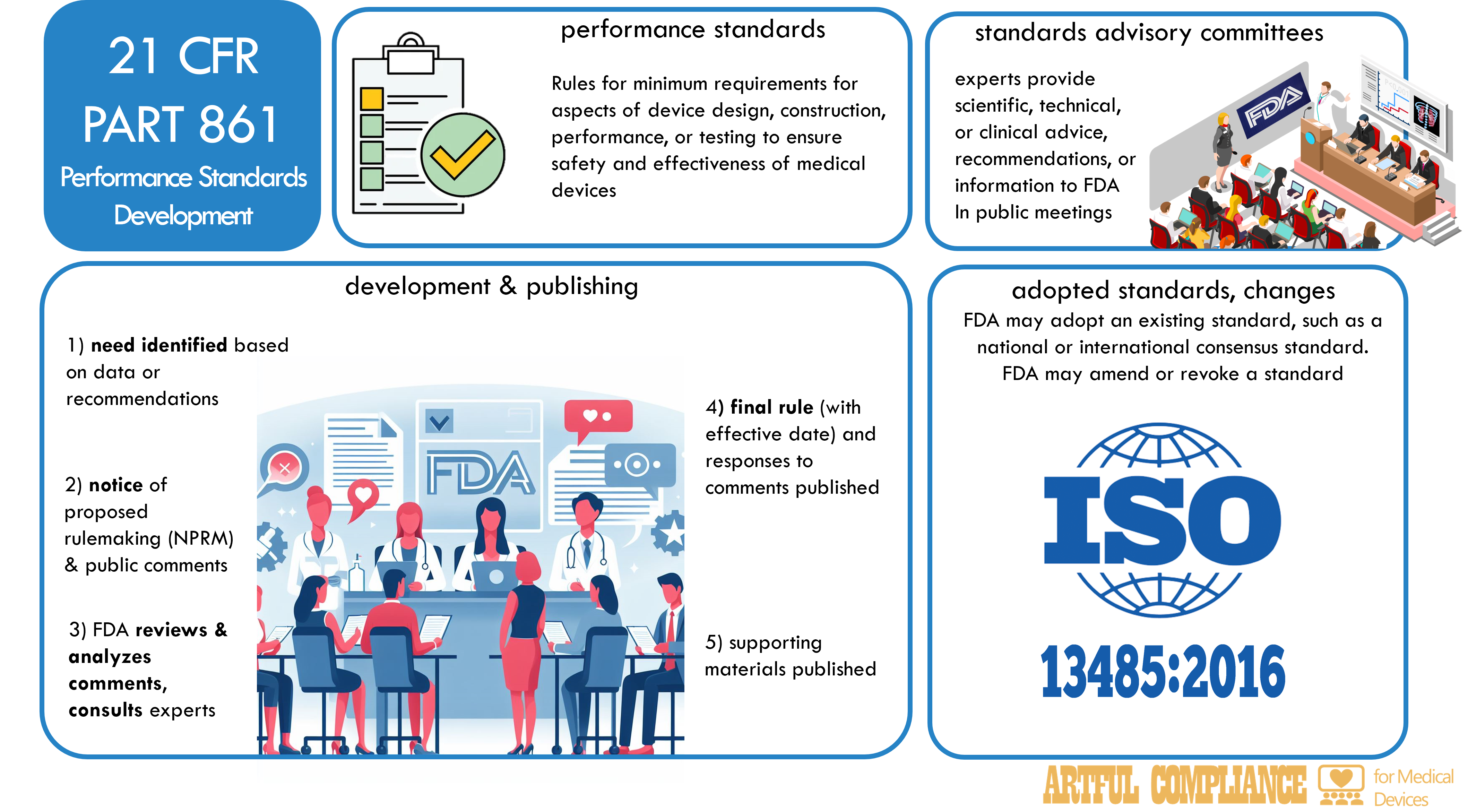
One of the ways that the FDA ensures the safety and effectiveness of medical devices is by establishing performance standards, which are rules that specify the minimum requirements for certain aspects of device design, construction, performance, or testing.
This regulation describes the procedures for developing, publishing, and revising performance standards, and the role of standards advisory committees in advising the FDA on technical matters related to performance standards. These procedures ensure that performance standards are based on sound scientific, technical, or clinical data, and that they provide reasonable assurance of the safety and effectiveness of medical devices.
Content of Standards
Performance standards established by the FDA include the following elements:
- The purpose and scope of the standard, including the device or devices to which it applies, the risk or risks that it addresses, and the intended benefit to public health.
- A description of the performance characteristics or requirements that the device or devices must meet, such as specifications, test methods, limits, or tolerances.
- The effective date of the standard, and any transitional or compliance periods for manufacturers, importers, or distributors of the device or devices.
- As applicable, any labeling, recordkeeping, reporting, or postmarket surveillance requirements.
Development and Publication
To develop and publish a performance standard, the FDA…
- identifies a need for a performance standard for a device or devices, based on scientific, technical, or clinical data, or on recommendations from standards advisory committees, other federal agencies, or external stakeholders.
- publishes a notice of proposed rulemaking (NPRM) in the Federal Register, which announces the FDA’s intention to establish a performance standard, and invites public comments on the proposed standard.
- reviews and analyzes the public comments, and consults with standards advisory committees, other federal agencies, or external stakeholders, as appropriate.
- publishes a final rule in the Federal Register, which establishes the performance standard, and responds to the public comments on the proposed standard.
- publishes a notice of availability in the Federal Register, which announces the availability of any guidance documents, test methods, or other supporting materials related to the performance standard.
Use of Existing Standards
In some cases, the FDA may adopt an existing standard, such as a national or international consensus standard, as a proposed or final performance standard for a device or devices, if the FDA determines that the existing standard is appropriate and adequate to provide reasonable assurance of the safety and effectiveness of the device or devices. For instance, the FDA is considering adoption of ISO 13485, an international standard for quality systems for medical devices. In such cases, the FDA will follow the same procedures for development and publication as described above, but will also include a reference to the existing standard, and an explanation of any differences or modifications that the FDA made to the existing standard.
Changes to Standards and Effective Dates
The FDA may amend or revoke a performance standard for a device or devices, if the FDA determines that the standard is no longer appropriate or adequate to provide reasonable assurance of the safety and effectiveness of the device or devices, or if the FDA finds new scientific, technical, or clinical data that warrant a change to the standard. The FDA will follow the same procedures for development and publication as described above, but will also include a statement of the reasons for the amendment or revocation, and the effective date of the change.
The effective date of a performance standard is the date on which the standard becomes legally binding for the device or devices to which it applies. The FDA may specify different effective dates for different provisions of the standard, or for different types or classes of devices. The FDA may also provide transitional or compliance periods for manufacturers, importers, or distributors of the device or devices, to allow them time to meet the requirements of the standard.
Standards Advisory Committees
Standards advisory committees are groups of experts that advise the FDA on technical matters related to performance standards for medical devices. The FDA may establish one or more standards advisory committees for a specific device or devices, or for a general area of device regulation. The members of the standards advisory committees are appointed by the FDA, and may include representatives of scientific, professional, industry, or consumer organizations, as well as federal employees or consultants.
The role of the standards advisory committees is to provide the FDA with scientific, technical, or clinical advice, recommendations, or information on the development, amendment, or revocation of performance standards, or on the use of existing standards. The FDA may consult with the standards advisory committees at any stage of the performance standards development process, and may request them to review and comment on proposed or final standards, or on any supporting materials related to the standards.
The standards advisory committees operate in accordance with the Federal Advisory Committee Act, which requires that their meetings be open to the public, and that their records and reports be available for public inspection.


Recent Comments